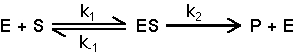
Experiment 3.2 Protease Kinetics (version: 02/09/00)
Background
Kinetics, the study of rates of chemical reactions, allows one to determine detailed mechanistic steps of chemical reactions. One can determine affinities of substrates, products and inhibitors for an enzyme, catalytic mechanisms, inhibition mechanisms, roles in coupled, sequential reactions, etc. In one of the simplest enzymatic mechanisms, an enzyme (E) and a substrate (S) combine to form an enzyme-substrate complex (ES), which decomposes to product (P) and enzyme.
One can simplify the rate equation by making several assumptions.
Assumption 1. Assume that k-1>> k2. If this assumption is correct, the first step of the reaction is essentially in equilibrium. The second step is very slow compared to the first step.
Assumption 2. Assume steady state. The steady state assumption is that [ES] is constant. This assumption is true when the rates of formation and decomposition of ES are the same. Steady state is not equivalent to equilibrium.
The resulting Michaelis-Menten equation is:
Vo = Vmax [S] / ( Km + [S] )
or
1/Vo = (Km/Vmax)1/[S] + 1/Vmax
Vo is the initial velocity of the reaction,
Vmax = k2[Etot], is the maximal
velocity of the reaction ,
[S] = substrate concentration,
Km = (k-1 + k2
) / k1. Km is the Michaelis Constant, the substrate
concentration at which the reaction velocity is half of the maximal velocity.
k2 is also known as kcat, the 'turnover
number', which is the number is reaction processes per unit time, per enzyme.
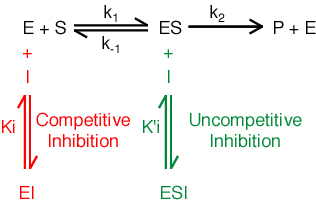
Inhibition. An inhibitor reduces the activity of an enzyme. Many important drugs and pharmaceutical agents are enzyme inhibitors. Agents that compete with substrate for binding to the enzyme are known as competitive inhibitors. Competitive inhibitors are characterized by an inhibition constant Ki.
Ki=[E][I]/[EI]
Agents that bind to the enzyme-substrate complex are known as un-competitive inhibitors.
One of the goals of this laboratory is to determine kinetic parameters (Km and kcat) for a reaction catalyzed by a protease. The mechanism and class of a serine protease are essentially independent of the specificity. For example both a-chymotrypsin and trypsin are serine proteases, with nucleophilic serine residues and conserved catalytic triads. Yet the S1 specificity of a-chymotrypsin differs from that of trypsin; a-chymotrypsin has S1 specificity for bulky aromatic residues (phenylalanine) while trypsin has S1 specificity for basic residues (arginine and lysine).
The Assay. You will determine kinetic parameters for the hydrolysis of the substrate N-alpha-benzoyloxycarbonyl-L-lysine thio-benzyl ester (CBZ-K-TBE). This substrate is not a peptide and is not a substrate for trypsin in vivo, but has several advantages for our in vitro assay. Enzymatic hydrolysis of this substrate occurs by the following scheme.
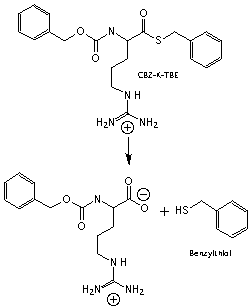
Figure 3: Hydrolysis of the substrate CBZ-K-TBE. Trypsin hydrolyzes CBZ-K-TBE into two products, including benzylthiol.
Neither of these products has a characteristic visible absorption. To visualize the reaction products, excess 5,5'-dithio-bis-2-nitrobenzoic acid (Ellman's reagent) is added to the reaction mixture, causing a second reaction to occur immediately after the first.
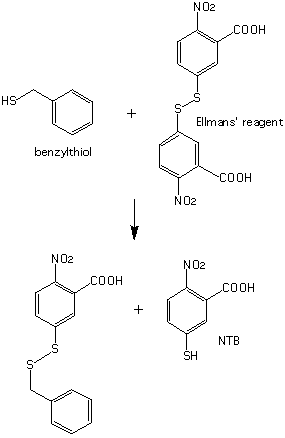
Figure 4: Reaction of benzylthiol with Ellman's reagent. The products include 2-nitro-5-thio-benzoic acid (NTB), a strongly colored species.
The highly chromophoric species 2-nitro-5-thio-benzoic acid (NTB), has a lambda max at 410 nm (e = 14173 AU/M). Each mole of benzylthiol produced enzymatically by trypsin forms one mole of NTB. The rate of this reaction is very fast compared with the rate of the enzymatic reaction. Therefore the rate of formation of NTB is essentially identical to the rate of hydrolysis of the ester (CBZ-K-TBE substrate). In addition, you will study inhibition of trypsin by small molecular weight compounds.
Prelab (top=>)
(to be performed before you arrive for lab)
a) Read Voet & Voet, Chapters 12-1, 12-2, 13, 14-3 and this handout.
b) Write a brief synopsis of the procedure in point form.
c) In your notebook construct a table that is ten columns by twelve rows. Label it Table 1. Label the columns:
| # | Vbuffer | V Ell's reagent | Vinhib | VDMSO | Vsub |
stock |
[sub] | Venzyme/HCl | AU/sec |
Partially fill out the table.
1) # is the row number (1-12).2) Vbuffer is 1700 microliters (1.7 ml) for all 12 rows.
3) VEll's reagent is 5 microliters (0.005 ml) for all rows.
4) Vinhib is 0 microliters for all rows.
5) VDMSO is100 microliters for all rows.
6) Vsub (volume substrate) and relative stock concentrations are:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
* A 0.1X substrate stock solution is one tenth as concentrated as a 1X stock solution. Make a 0.1X stock solution by a ten-fold dilution of a 1X stock solution.
** A 0.01X substrate solution is one one hundredth as concentrated as a 1X solution. Make a 0.01X solution by a ten-fold dilution of a 0.1 X solution
7) During or after lab calculate the concentration of the substrate ([substrate]).d) Make two additional tables (Tables 2 and 3) that are identical to Table 1 except that the VDMSO and Vinhib entries are left blank. You will fill in these values as you do the lab.8) Venzyme/HCl is 25 microliters for all rows.
9) Fill in the AU/sec values during the experiment.
e) Answer the following questions:
1) In three sentences, describe the advantage of using a double beam spectrophotometer for this experiment.Materials and Methods (top=>)2) Give the order in which agents are added to a cuvette.
The following reagents are required for the enzyme assays:
|
|
(°C) |
|
|
|
|
(1x) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(concentrated) |
|
(on ice) |
|
|
|
(dilute) |
|
(on ice) |
|
2 x 10-6 M |
|
Reagent |
|
|
|
|
|
|
|
|
CaCl2 |
100 mM, 10 mM |
|
|
|
|
|
|
* A 1x substrate solution is ten times as concentrated as a 0.1x solution.
** A 1x substrate solution is one hundred times as concentrated as a 0.01x solution.
*** These solutions will be provided by the TA. Use the dilute solution (Enzyme B) for your kinetic assays and the concentrated solution (Enzyme A) for determining substrate concentration.
Required Supplies and Instrumentation (top=>)
10 microliter pipette tips (white)Procedure (top=>)200 microliter pipette tips (yellow)
1000 microliter pipette tips (blue)
1-10 microliter pipetteman
10-100 or 20-200 microliter pipetteman
1000 microliter pipetteman
100 precut 1" by 1 " squares of parafilm
1 box (100) disposable cuvettes
UV-vis spectrophotometer
Ice bucket
1) Set-up the Shimadzu
UV-1601 spectrophotometer.
The Shimadzu instrument (UV-1601) used in this laboratory
is a double-beam spectrophotometer. Open up the top cover and examine the
cuvette holder. The back holder is for the Reference (R) cuvette, the forward
holder is for the sample (S) cuvette. A double beam spectrophotometer calculates
the absorbance difference between S and R. In this lab, a small but significant
increase of the absorbance will be caused by non-enzymatic hydrolysis of
substrate, catalyzed by water. If the sample in the R cuvette is idential
to that of the S cuvette, except that the R cuvette contains no enzyme,
the increase in absorbance due to non-enzymatic substrate hydrolysis is
automatically subtracted out.
a) Turn on spectrophotometer; the switch is on the left.2) Determine the substrate stock concentrations. (top=>)
b) Allow machine to go through self check (takes about 1 minute).
a) Zero the Spectrophotometer. Put identical solutions in the R and S cuvettes. To R and S cuvettes, add 1700 microliters of buffer plus 5 microliters Ellman's Reagent. Then cover both cuvettes with parafilm and invert three times. Place the R and S cuvettes in the spectrophotometer.
b) Upon starting up the spectrophotometer, the "Mode" menu will appear with eight choices. Press "Photometrics". Set the wavelength to 410 nm: press the GOTOWL button, type 410 nm, and press enter. Throughout this lab, ensure that the spec remains at this wavelength. Autozero the instrument. Note the absorbance at 410 nm after it stops changing with time. It should read very close to zero. Remove both cuvettes from the spectrophotometer.
c) Determine the absorbance after 100% reaction of a known volume of substrate. Remove the cuvettes from the spectrophotometer. Do not discard the contents.
i) To the R cuvette, add 50 microliters of H203) Enzyme Assays with no Inhibitor. (top=>)ii) To the S cuvette, add 25 microliters trypsin solution A (concentrated) plus 25 microliters of substrate (diluted 1:10 in water),
iii) Cover both cuvettes with parafilm, invert three times and place them in the spectrophotometer. Record the absorbance at 410 nm after it stops increasing with time. Record the absorbance again to obtain a duplicate value. If the results are not between 0.9 and 1.1 AU please confer with your the TA.
To determine the kinetic parameters Km, Vmax and kcat for hydrolysis of CBZ-K-TBE by trypsin, you will determine the initial rates of the reaction (Vo) for a range of substrate concentrations [S]. The goal is to use the same buffer, pH, [DMSO], [substrate], etc., in the reference (R) and sample (S) cuvettes. The only difference between the two cuvettes is that S will contain the enzyme and R will not.
a) From the "Mode" menu on the spectrophotometer press "Kinetics".
Set the following parameters (To change a parameter, press the number and change the value accordingly, to go back to a previous menu, press "return").
i) Meas. mode: ABSc) Ensure that the wavelength is 410 nm.ii) Meas. Time: 30 secs, Lag Time: 2 secs, Rate time: 5 secs
iii) Factor: 1.000
iv) Rec. range: 0 to 0.2 AU
v) Temp. control: none
vi) Time scale: sec
b) Line up two rows of 12 cuvettes each. Label one row 1S1, 2S1, 3S1... and the other row 1R1, 2R1, 3R1... (do not write on the optical surface).
To all 24 cuvettes add:
i) buffer (1700 microliters)To make the first measurement addii) Ellman's reagent (5microliters)
iii) DMSO (100 microliters)
iv) substrate to the 1R1 and 1S1 cuvettesImmediately after the cuvettes are in place, close the lid and press auto zero, then start. The goal is to obtain the initial velocity (Vo). So time becomes critical as soon as the enzyme is added to the S cuvette. You should be able to add the enzyme and begin making measurements within several seconds. Observe the AU versus time graph to ensure that it is close to linear. After a measurement is complete, press Data/List to view the reaction velocity (AU/min). The reaction velocity in absorbance units per minute, and will be printed out for each run. Record the values in your notebook, and take the printed table with you.v) 1 mM HCl to the 1R1 cuvette only. Then mix by placing a piece of parafilm over the mouth of the cuvette and inverting four times. Place the cuvette in the back cuvette holder in the spectrophotometer.
vi) enzyme solution B to the 1S1 cuvette only. Then mix and place the cuvette in the forward holder.
After completing the first measurement repeat iv, v, vi for the 2R1 and 2S1 cuvettes, then 3R1 and 3S1, etc.
4) Enzyme Assays with Inhibitor (determine the Ki of an unknown inhibitor). (top=>)
One of your goals is to determine the type and the Ki of your unknown inhibitor. You will be given an inhibitor solution of (known) concentration, but will need to dilute it. First determine two appropriate volumes and concentrations of the inhibitors to use. Determine the velocity under the same conditions as 1S1 but in the presence of the inhibitor. Adjust the amount of inhibitor to achieve (1) 10-15% and, (2) 20-25%, of the velocity in the absence of inhibitor. So if 1S1 gives a Vo of 0.30 AU/minute add enough inhibitor to give (1) 0.26 OD/min and (2) 0.23 OD/min. It may be neccessary to further dilute some of the inhibitors in DMSO. If you do this make a record of it. You need to know the inhibitor concentration to calculate Ki. Since the inhibitor is dissolved in 100% DMSO you must ensure that the total volume of DMSO added to the R and S cuvettes is always the same (100 mL). If you add 10 mL of inhibitor solution, add 90 mL of additional DMSO. If you add 30 mL of inhibitor, add 70 mL of additional DMSO. For each of the two inhibitor concentrations, determine the rates of reaction for differing substrate concentrations as in Section 3, above.You will be given one unknown inhibitor from the following list. Inhibitor stock solutions are 100 mM.
4-chlorobenzylamine (FW:141.60 grams/mole) Irritant, combustible liquidCaution: WEAR GLOVES WHEN HANDLING THE INHIBITORS. You should not dilute any of the inhibitor solutions to less than 20% of its original concentration. Dump inhibitor waste in the glass bottle marked "Inhibitor Waste".
4-(Aminomethyl)benzoic acid (FW 151.17) (Irritant)
Benzamidine hydrochloride (FW 156.61) (Irritant)
Benzylamine (FW 140.61) (Corrosive, lachrymator, combustible)
4-methoxybenzylamine (FW 137.18) (Corrosive)
4-aminobenzamide (FW 136.15) (Irritant)
4-aminobenzylamine (FW 122.17) (Corrosive)
a) For the first inhibitor concentration (10-15% inhibition), line up two rows of 12 cuvettes each. Label one row 1S2, 2S2, 3S2... and the other row 1R2, 2R2, 3R2... (do not write on the optical surface). As given for Table 2: to all 24 cuvettes add:
i) bufferTo make the first measurement addii) Ellman's reagent
iii) inhibitor
iv) DMSO (VDMSO = 100ml - Vinhib)
v) substrate to the 1R2 and 1S2 cuvettesImmediately after the cuvettes are in place in the spectrophotometer, close the lid, press auto zero and start. Make sure that absorbance versus time is linear. Press Data/List to recover the reaction velocity (AU/min), and write this number down.vi) 1 mM HCl to the 1R2 cuvette only. [Mix and place in spectrophotometer.]
vii) enzyme solution B to the 1S1 cuvette only [Mix and place in spectrophotometer.]
viii) After completing the first measurement repeat v, vi, vii for the 2R2 and 2S2 cuvettes, then the 3R2 and 3S2, etc.b) For the second inhibitor concentration (20-25% inhibition), line up two rows of 12 cuvettes each. Label one row 1S3, 2S3, 3S3... and the other row 1R3, 2R3, 3R3... Then proceed as above, except with a different inhibitor concentration.
Results and Discussion (top=>)
1) Determine the substrate stock concentration (Ms) with Beer's Law. To calculate the concentration in the cell use A = (e)(pathlength)(Mc), where A is the absorbance, e is the extinction coefficient and Mc is the concentration in the cell. To calculate the concentration of the stock solution use Ms = McVc/Vs where Ms is the concentration of the stock solution, Vc is the total volume of solution in the cell, and Vs is the volume of stock solution that you have added to the cell. Show this calculation.
2) Determine the concentration of the inhibitor in the cuvette for each volume of added inhibitor. The inhibitor stock concentration is given. Show an example of this calculation. If you use a a spreadsheet, include a copy.
3) Determine the Km of substrate.
Construct a 5 x12 table with the following column titles. Vo is the initial velocity.
|
|
|
|
|
|
Plot [substrate] versus initial velocity. Fit the curve in the best way you can as in Voet, Figure 13-8 (page 352). Estimate Vmax and Km from this plot. Also plot (1/[substrate]) versus (1/velocity). This is a Lineweaver - Burk plot, known as a double reciprocal plot. Fit a line into the data with a linear least sequares program, Voet and Voet, Figure 13-9 (page 353). Estimate Vmax and Km from this plot. Calculate kcat.
3) Determine the Ki of Inhibitor.
Determine the concentration of inhibitor for each experiment from the known stock concentration and dilution factor. Show an example of one of these calculations. For each inhibitor concentration, construct 5 x12 tables with the following column titles.
|
|
|
|
|
|
You should have three of these tables. Plot all (1/[substrate]) versus (1/Vo) lines on the same graph Determine the Ki of the inhibitor as described in Voet and Voet, pages 356-360.
4) Write a DISCUSSION as described in the syllabus.
5) What is the significance of Km?
6) What is the significance of kcat?
7) What is the significance of kcat/km?
8) What do your 1/Vo versus 1/[S] graphs say about the mechanism of your inhibitor?
9) What is the significance of Ki?
References (top=>)
1) Neurath, H. "Evolution of proteolytic enzymes." Science 224, 350-357 (1984).
2) Voet, D. Biochemistry, 2nd Ed., John Wiley and Sons (1995)
3) Creighton, T.E., Proteins, 2nd Ed., W. H. Freeman and Co. (1993)
4) Schechter, I., and Berger, A. "On the size of the active site in proteases. I. Papain." Biochem. Biophys. Res. Comm., 27, 157-162.