2) Heat the mixture on a stirring hot plate until the agarose dissolves. Do not let it boil.
3) Assemble the gel casting tray and comb. The comb should not touch the bottom of the tray.
4) When the agarose solution has cooled to about 50°C (warm but not scalding - cool it in a water bath to save time), pour the agarose solution carefully into the mold, ensuring that no bubbles get into the gel. Do not pour the agarose solution into the casting tray if the temperature is greater than 50°C, as the casting tray will crack or warp. Rinse the flask immediately after filling the tray.
5) Allow the gel to cool. It will solidify and become slightly opaque within 20 to 30 minutes.
6) Carefully remove the comb by lifting it gently at one end, tilting the comb as it comes out. Pulling the comb straight up creates a vacuum in the wells, which tends to lift the whole gel out of the tray.
7) Lift the gel and plastic running tray from the casting tray and remove any agarose adhering to the bottom of the tray. Place the gel on the center platform of the running unit.
8) Add TAE running buffer to the reservoirs of the gel aparatus. Submerge the gel by around a half a centimeter. Ensure that the wells are submerged and filled.
9) Prepare the DNA samples, including the size markers (length standards), for loading on the gel. Add 2 microliters of loading buffer to each sample. Mix well.
10) Sketch the gel, with numbered lanes and corresponding contents, in your notebook. Slowly load each sample into a well with a P-20 Pipetman, which is easier to control than a P-200. Place the pipet tip containing the sample near the bottom of the well. Be careful not to puncture the well. Slowly expel the sample and simultaneously raise the tip to the top of the well. If you leave the tip at the bottom of the well, there may not be sufficient volume for the sample. Do not use the "blow out" feature of the pipet, which will expel an air bubble and clear the sample out of the well. Do not stir the contents of the well.
11) Once all the samples are loaded place the cover on the gel apparatus. Connect the leads so that the red (positive) lead is at the end of the gel to which the DNA will migrate and the black (negative) lead is at the end of the gel containing the wells. Turn on the power supply and set according to the instructor's guidelines. Check the gel after a few minutes. If the dye migrates in the wrong direction, turn off the power supply and switch the leads. Run at 85 volts. The larger power supplies have a dial or buttons for adjusting the voltage. Adjust to 50-70 volts. More than one gel may be run on a power supply at once. Keep voltage constant. DO NOT REMOVE THE LID FROM THE GEL APPARATUS WITHOUT DISCONNECTING THE POWER LEADS. ELECTRICAL SHOCK MAY RESULT.
12) When the blue tracking dye (which runs in these gels along with a DNA fragment of about 200-400 bp) has migrated about 75% of the distance to the end of the gel (usually 30-60 minutes), turn off the power supply and disconnect the power leads.
13) Stain the gel with ethidium bromide. Ethidium bromide is a carcinogen and must be handled with gloves. The gel must be handled with gloves from this step on. Add about 100 ml of water and 20 ul of ethidium bromide (10 mg/ml stock) to the tray. Allow the gel to stain for around 5 minutes. Rock the tray a couple of times during the soaking.
14) Visualize the DNA with UV light. (UV light damages skin and eyes, wear a protective mask.) Place the gel on the light box. If the DNA is not visible, stain the gel for 5 additional minutes then destain the gel by soaking in dd water for 10 minutes. Destaining will remove the background glow.
15) Photograph the gel under the UV light, as instructed by a TA. If you have a clear picture, place the gel in the trash.
16) Carefully pour the ethidium bromide solution into the appropriate waste container.
17) Rinse the light box and tray with dd water and dry them with paper towels.
DNA Size Markers: Lambda DNA-BstEII Digest (NEB #301-4L 750 µg $200)
These markers are made by a BstEII digest of lambda DNA (cI857ind1 Sam 7) to yield 14 fragments suitable for use as molecular weight standards for agarose gel electrophoresis.
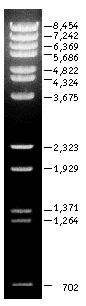 Lambda
DNA-BstE II Digest visualized by ethidium bromide staining. 1.0% agarose
gel.
Lambda
DNA-BstE II Digest visualized by ethidium bromide staining. 1.0% agarose
gel.