4581 =>
(revision 1/05/2001)
SDS Page Gel Electrophoresis
PAGE
Polyacrylamide gel electrophoresis (PAGE) is probably the most common analytical technique used to separate and characterize proteins. A solution of acrylamide and bisacrylamide is polymerized. Acrylamide alone forms linear polymers. The bisacrylamide introduces crosslinks between polyacrylamide chains. The 'pore size' is determined by the ratio of acrylamide to bisacrylamide, and by the concentration of acrylamide. A high ratio of bisacrylamide to acrylamide and a high acrylamide concentration cause low electrophoretic mobility. Polymerization of acrylamide and bisacrylamide monomers is induced by ammonium persulfate (APS), which spontaneously decomposes to form free radicals. TEMED, a free radical stabilizer, is generally included to promote polymerization.
SDS PAGE
Sodium dodecyl sulfate (SDS) is an amphipathic detergent. It has an anionic headgroup and a lipophilic tail. It binds non-covalently to proteins, with a stoichiometry of around one SDS molecule per two amino acids. SDS causes proteins to denature and dissassociate from each other (excluding covalent cross-linking). It also confers negative charge. In the presence of SDS, the intrinsic charge of a protein is masked. During SDS PAGE, all proteins migrate toward the anode (the positively charged electrode). SDS-treated proteins have very similar charge-to-mass ratios, and similar shapes. During PAGE, the rate of migration of SDS-treated proteins is effectively determined by molecular weight.
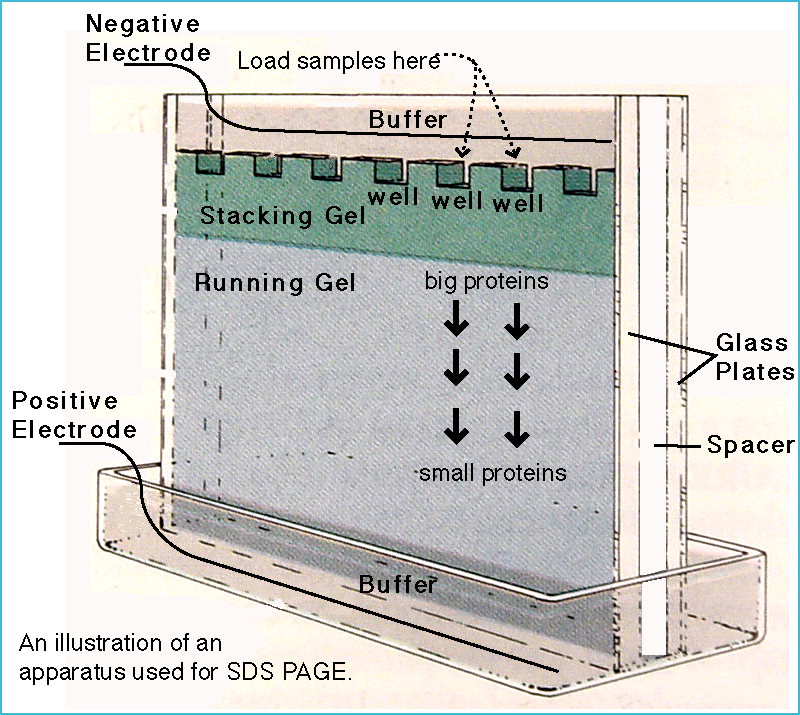
Below is an example of the procedure for performing discontinuous SDS-PAGE with a 14% separating gel and a 5% stacking gel.
Materials
PAGE Rigs including glass plates (10 x 20 cm), spacers, comb, and clamps
Power supply
Protein sample
Bio-Rad Laemmli Sample Buffer (contains SDS and either sucrose or glycerol)
2-Mercaptoethanol (reduces disulfide bonds, disrupts protein cross-links)
MW Markers (already prepared in sample buffer)
Gel Cassette Assembly (Bio-Rad Mini Protean 3)
- Clean and completely dry the glass plates, combs, and any other pertinent materials.
- Place a short plate on top of a spacer plate. Insert both plates into the green casting frame on a flat surface. Be sure that the "legs" of the casting frame are down. Clamp the casting frame and check that the plates are level on the bottom.
- Put the casting frame into the casting stand.
Preparation of the Gel
- Combine all reagents except the TEMED for the 14% separating gel.
1.184 milliliters deionized water
1.96 milliliters 30% acrylamide/Bis (Warning: Acrylamide is a neurotoxin. Use gloves, do not ingest.)
1.0 milliliters 1.5 M Tris, pH 8.8
21 microliters 20% SDS
12 microliters 10% ammonium persulfate
18 microliters TEMED, pH 8.9
- When ready to pour the gel, quickly add the TEMED, mix using a Pasteur pipette, and transfer the separating gel solution between the glass plates in the casting chamber to about 3/4 inch below the short plate.
- A small layer of butanol can be added on top of the gel prior to polymerization to straighten the level of the gel and remove unwanted air bubbles that may be present. Butanol will not mix with the aqueous separating gel solution. Once the gel has polymerized, the butanol can be removed by absorption with Kimwipes or filter paper. Rinse the top layer of the gel with dI water prior to pouring the stacking gel.
- Insert the well forming comb into the opening between the glass plates.
- Combine all reagents except the TEMED for the 5% stacking gel.
0.4 milliliters deionized water
1.8 milliliters 30% acrylamide/Bis
0.25 milliliters 0.5 M Tris, pH 6.8
12 microliters 20% SDS
17 microliters 10% ammonium persulfate
5 microliters TEMED
- When ready to pour the gel, quickly add the TEMED, mix using a Pasteur pipette, and transfer the stacking gel solution between the glass plates in the casting chamber.
- Both the separating and stacking gels should polymerize within six minutes.
- Once the stacking gel has polymerized, the comb can be gently removed. The polymerized gel between the short plate and spacer plate forms the "gel cassette".
Sample Preparation
- Place some water in a 600 mL beaker and leave on a hot plate to boil. (This can take 15 minutes or more.)
- Meanwhile, add 50 mL of 2-mercaptoethanol to 950 mL of Laemmli sample buffer.
- Combine 10 mL of each protein sample with 20 mL of Laemmli sample buffer plus 2-mercaptoethanol in microcentrifuge tubes.
- In separate tubes, aliquot 10 mL of MW marker. (MW markers are already prepared in Laemmli sample buffer.)
- Boil the samples for no more than 5 minutes to fully denature the proteins. Leave the samples at room temperature until ready to load onto the gel.
Electrophoresis
- Remove the gel cassette from the casting stand and place it in the electrode assembly with the short plate on the inside.
- Slide the electrode assembly (with the gel cassette) into the clamping frame. Press down on the electrode assembly while clamping the frame to secure the electrode assembly. This step is important to minimize potential leakage during the electrophoresis experiment.
- Pour some 1X electrophoresis buffer into the opening of the casting frame between the gel cassettes. Add enough buffer to fill the wells of the gel. Use a gel loading tip to pipette some buffer into each well to ensure cleanliness.
- When all wells are sufficiently cleaned, slowly pipette no more than 30 mL of denatured sample or MW marker into each well. A yellow guide can be placed on top of the electrode assembly to aid in loading the gel.
- When the gel has been loaded, lower the clamping frame into the electrophoresis tank.
- Fill the region outside of the frame with 1X electrophoresis buffer.
- Cover the tank with the lid aligning the electrodes (black or red) appropriately.
- Connect the electrophoresis tank to the power supply.
- Allow the samples to run at 30 mA until the dye front reaches the bottom of the gel. This can take as long as 1 hour.
- When electrophoresis is complete, turn off the power supply and disassemble the apparatus.