Interacciones Moleculares
(Interacciones no covalentes)
Loren Dean Williams
Escuela de Química y Bioquímica, Georgia Tech,
Atlanta, Georgia, USA
5/18/2024
Traducido por:
Ignacio Amigo. Science writer. Madrid, Spain.
Celia Blanco. Department of Chemical and Biomolecular Engineering, UCLA Samueli School of Engineering, University of California, Los Angeles. celiablanco@ucla.edu
Isaac Gállego. MRC Laboratory of Molecular Biology, Cambridge, UK. igallego@mrc-lmb.cam.ac.uk
Índice de Contenido

Las interacciones moleculares son fuerzas atractivas o repulsivas entre moléculas y entre átomos no enlazados. Las interacciones moleculares son importantes en diversos campos, como el estudio del plegamiento de proteínas, el diseño de fármacos, la ciencia de materiales, el desarrollo de sensores, la nanotecnología, los procesos de separación y la investigación sobre los orígenes de la vida. A las interacciones moleculares también se las conoce como ‘interacciones no covalentes’ o ‘interacciones intermoleculares’.
Las interacciones moleculares no son enlaces. Los enlaces mantienen los átomos juntos en las moléculas. Una molécula es un conjunto de átomos que se asocia lo suficientemente fuerte como para no disociarse o perder su estructura cuando interactúa con su entorno. A temperatura ambiente, dos átomos de nitrógeno forman un enlace (N2). Dos átomos de argón no. Los enlaces se rompen y se forman durante las reacciones químicas. En la reacción química en la que el fuego consume un trozo de papel, los enlaces de la celulosa se rompen mientras se forman enlaces de dióxido de carbono y agua. La entalpía de enlace es una magnitud termodinámica que cuantifica la energía total contenida en un enlace químico — es la energía necesaria para romper o formar enlaces. Las entalpías de enlace suelen ser del orden de 100 kcal/mol (400 kJ/mol), que es mucho mayor que RT a temperatura ambiente (donde R es la constante de los gases y T es la temperatura); los enlaces no se rompen a temperatura ambiente.
Los enlaces permanecen intactos cuando (a) el hielo se derrite, (b) el agua hierve, (c) el dióxido de carbono se sublima, (d) las proteínas se despliegan, (e) el ARN se despliega, (f) las cadenas de ADN se separan y (g) las membranas celulares se vuelven más fluidas. Estos procesos de fusión, ebullición, sublimación, despliegue, separación de hebras y cambio de fluidez implican alteraciones en las interacciones moleculares y no son reacciones químicas (no se rompen ni se forman enlaces). La entalpía de una interacción molecular entre un par de átomos no enlazados, es 1-10 kcal/mol (4-42 kJ/mol), que en el límite inferior es del orden de RT y en el límite superior es significativamente menor que un enlace covalente. Aunque individualmente son energías de carácter débil, acumulativamente las interacciones moleculares son significativas. Veamos algunos ejemplos:
Puntos de ebullición. Cuando una molécula pasa de la fase líquida a la fase gaseosa (como durante la ebullición), idealmente todas las interacciones moleculares se perturban. Los gases ideales son los ÚNICOS sistemas donde no hay interacciones moleculares. La diferencia en las temperaturas de ebullición es un buen indicador cualitativo de la intensidad de las interacciones moleculares en la fase líquida. Los líquidos con alto punto de ebullición tienen fuertes interacciones moleculares. El punto de ebullición del agua es cientos de grados mayor que el punto de ebullición del nitrógeno molecular, debido a que las interacciones moleculares en H2O (liq) son más fuertes que en N2 (liq). Las fuerzas entre las moléculas en H2O (liq) son mayores que las de N2 (liq).
Lennard-Jones. El potencial de Lennard-Jones (L-J) es una descripción empírica de las interacciones moleculares. Sin embargo, el potencial de L-J no incluye todas las interacciones moleculares. Las interacciones electrostáticas no están incluidas en el potencial L-J.
Estados nativos. En los sistemas biológicos (i) las proteínas se pliegan en estructuras globulares llamadas 'estados nativos', (ii) los ARN ribosómicos y de transferencia también se pliegan en estructuras globulares nativas, (iii) el ADN forma hélices de doble cadena, (iv) los fosfolípidos forman bicapas y (v) las proteínas se insertan en bicapas (para formar membranas) e interaccionan con ADN, ARN y otras proteínas. Estos estados nativos y relaciones moleculares se estabilizan mediante un gran numero de interacciones moleculares de elevada complejidad.
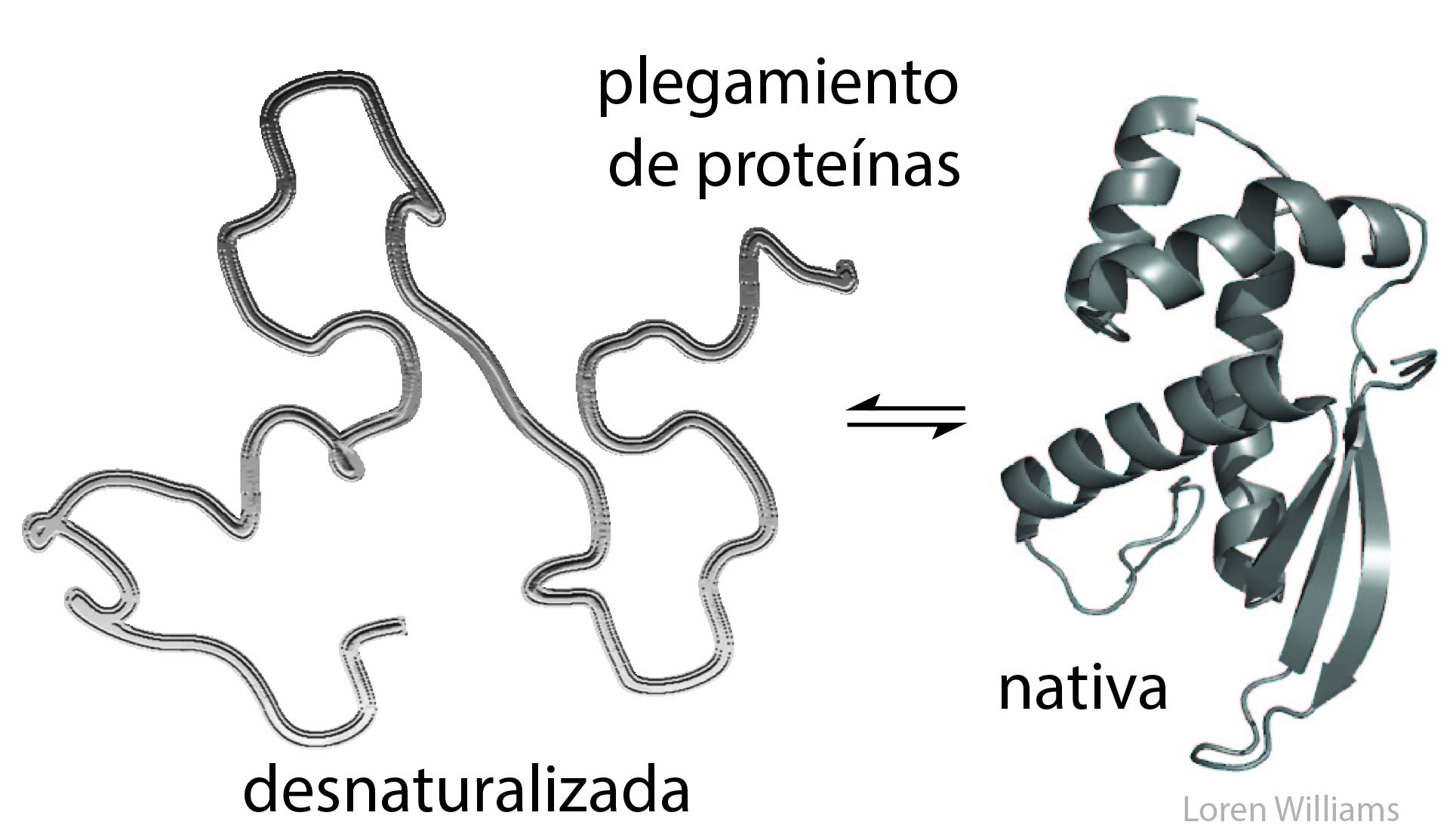
La figura 1 muestra el plegamiento de una proteína. El plegamiento de proteínas es la conversión de un estado desnaturalizado (un ovillo aleatorio) a un estado nativo. En el lado derecho, las flechas son hojas β y los tirabuzones son hélices α.
Estados desnaturalizados. Cuando se despliegan una proteína o un ARN (se desnaturalizan) o se separan dos cadenas de ADN (se deshibridan), o se desensambla un ribosoma, las regiones internas quedan expuestas al entorno, que en su mayoría está compuesto de agua e iones. Las interacciones moleculares dentro del estado o ensamblaje nativo se reemplazan por interacciones moleculares con entornos acuosos.
Cerca del punto de inflexión. Cuando se piensa en la estabilidad de un estado plegado (o un estado ensamblado), hay que tener en cuenta que las interacciones moleculares estabilizan tanto el estado plegado como el ovillo aleatorio (y el estado desensamblado). Un gran número de interacciones intramoleculares (dentro de una proteína) en estado nativo se oponen a otro gran número de interacciones intermoleculares en el estado desnaturalizado (con las moléculas de agua circundantes). Normalmente las macromoléculas y ensamblados biológicos son relativamente poco estables. Los sistemas biológicos en general se mantienen cerca del punto de inflexión, y pueden ser desplazados en direcciones opuestas por fuerzas muy intensas. Una pequeña perturbación puede inclinar la balanza del estado plegado al estado desplegado. Un pequeño cambio en el pH o en la temperatura, o una única mutación pueden desplegar una proteína. Los estados nativos tienen baja entropía conformacional. El ovillo aleatorio tiene una alta entropía conformacional.

¿Alguna vez has desnaturalizado una proteína (la has convertido del estado nativo al estado desnaturalizado)? Sí. Cuando calientas un huevo a unos 60°C, las proteínas de albúmina se desnaturalizan y agregan. No estás rompiendo enlaces cuando cocinas un huevo — estás cambiando y reorganizando las interacciones moleculares. La proteína agregada forma grandes conjuntos que dispersan la luz, dando al huevo una apariencia blanca. Cuando agregas jugo de limón a la leche, el pH se reduce y las proteínas se desnaturalizan y agregan. ¿Alguna vez has desnaturalizado el ADN? Sí, si has realizado una reacción en cadena por polimerasa (PCR).
Las interacciones moleculares fueron descubiertas por el científico holandés Johannes Diderik van der Waals, que fue el primero en darse cuenta de que las moléculas son pegajosas, un poco como las gominolas. Históricamente, la expresión 'interacción de van der Waals' se ha utilizado para referirse a fuerzas cohesivas (atracción entre similares), adhesivas (atracción entre diferentes) y/o a fuerzas repulsivas entre moléculas. Por ello, el término 'interacción de van der Waals' no es ni suficientemente informativo, ni descriptivo ni específico. Algunas personas lo usan para describir la totalidad de las interacciones moleculares (todas aquellas listadas en el Índice de Contenido), mientras que otras lo usan para describir algunos subconjuntos de interacciones moleculares. Aquí evitamos el término 'interacción de van der Waals', tanto por no estar bien definido como por no describir las interacciones de una manera adecuada desde un punto de vista físico.
Todas las interacciones moleculares son fundamentalmente de naturaleza electrostática y pueden describirse mediante alguna variación de la Ley de Coulomb. Sin embargo, reservamos el término 'interacción electrostática' para describir las interacciones entre especies cargadas formalmente. Las interacciones entre cargas parciales reciben otros nombres.
Hay muchas formas diferentes de analizar o clasificar las interacciones moleculares. Aquí usamos las categorías listadas en el Índice de Contenido por ser más claras, fáciles de entender y ser utilizadas ampliamente en la literatura.
Los átomos ocupan espacio. Si se intenta juntar dos átomos, se repelerán. Cuando dos átomos están muy juntos, los orbitales ocupados en las superficies de los átomos se superponen, causando repulsión electrostática entre los electrones de la superficie. Esta fuerza repulsiva entre los átomos actúa en un rango muy corto, pero es muy grande cuando las distancias son cortas.
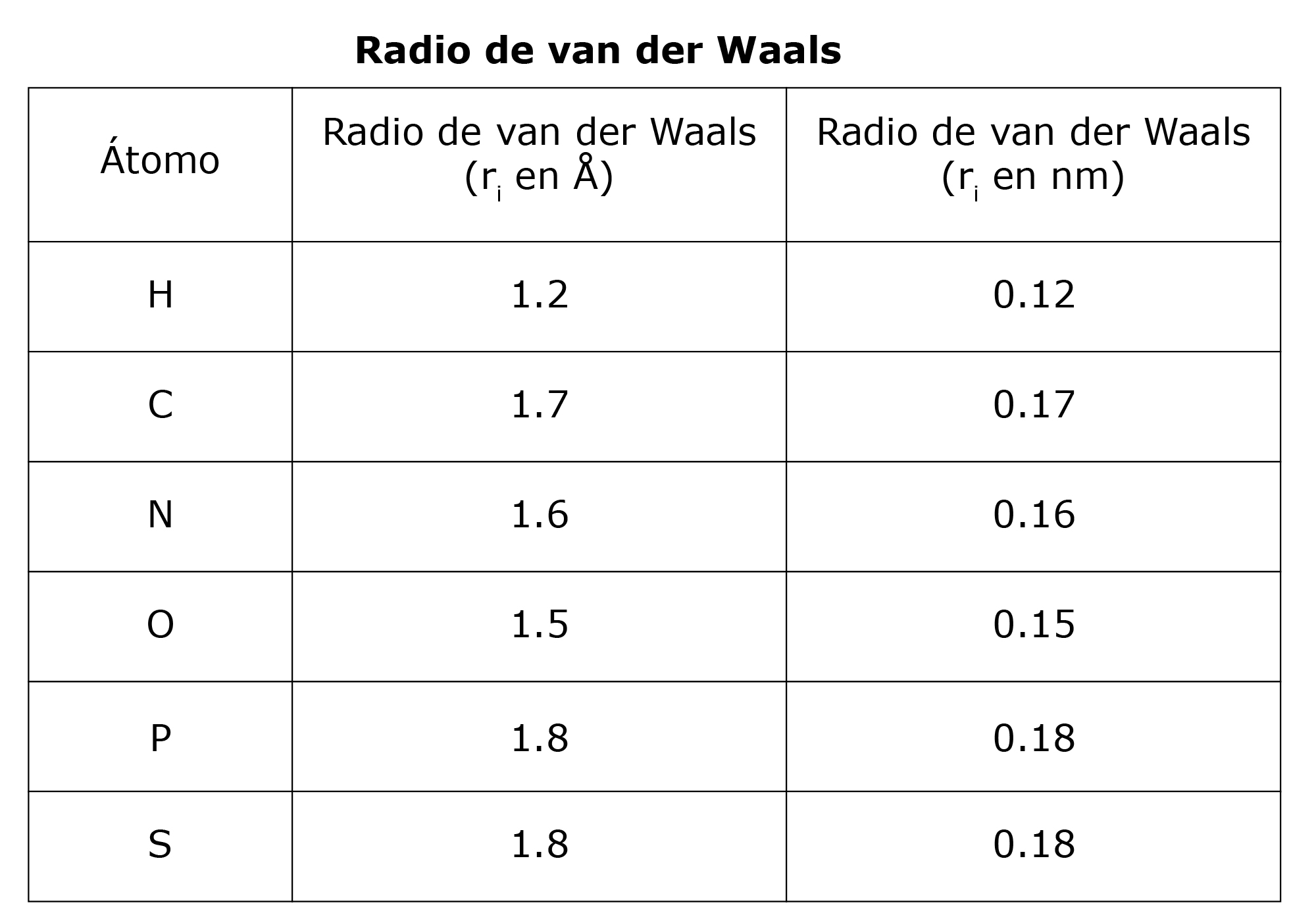
La energía repulsiva aumenta en función de (di/R)12, donde R es la distancia entre los átomos y di es el umbral de distancia por debajo del cual la energía se vuelve repulsiva. El valor de di depende del tipo de átomos que interactúan. Al tener una componente exponencial elevada, cuando R<di pequeñas disminuciones en R causan grandes aumentos en la repulsión. La repulsión de corto alcance solo importa cuando los átomos están muy cerca (R<di), pero a corta distancia dominan otras interacciones. Debido a que esta repulsión aumenta tan marcadamente a medida que disminuye la distancia, a menudo es útil considerar a los átomos como si fueran esferas sólidas, como bolas de billar muy pequeñas, con superficies duras (llamadas superficies de van der Waals) y radios bien definidos (llamados radios de van der Waals).
Cuando dos átomos se acercan, sus superficies de van der Waals hacen contacto cuando la distancia entre ellos es igual a la suma de sus radios de van der Waals. A esta distancia, la energía repulsiva incrementa repentinamente. La distancia más pequeña entre dos átomos no unidos es la suma de los radios de van der Waals de los dos átomos. Un átomo de azufre y un átomo de carbono no pueden estar más juntos que:
rS + rC = 1.8 + 1.7 = 3.5 Å.
Por supuesto, aquí estamos asumiendo que los enlaces no se forman. Cuando dos átomos forman un enlace, se acercan mucho y se viola su radio y superficie de van der Waals.
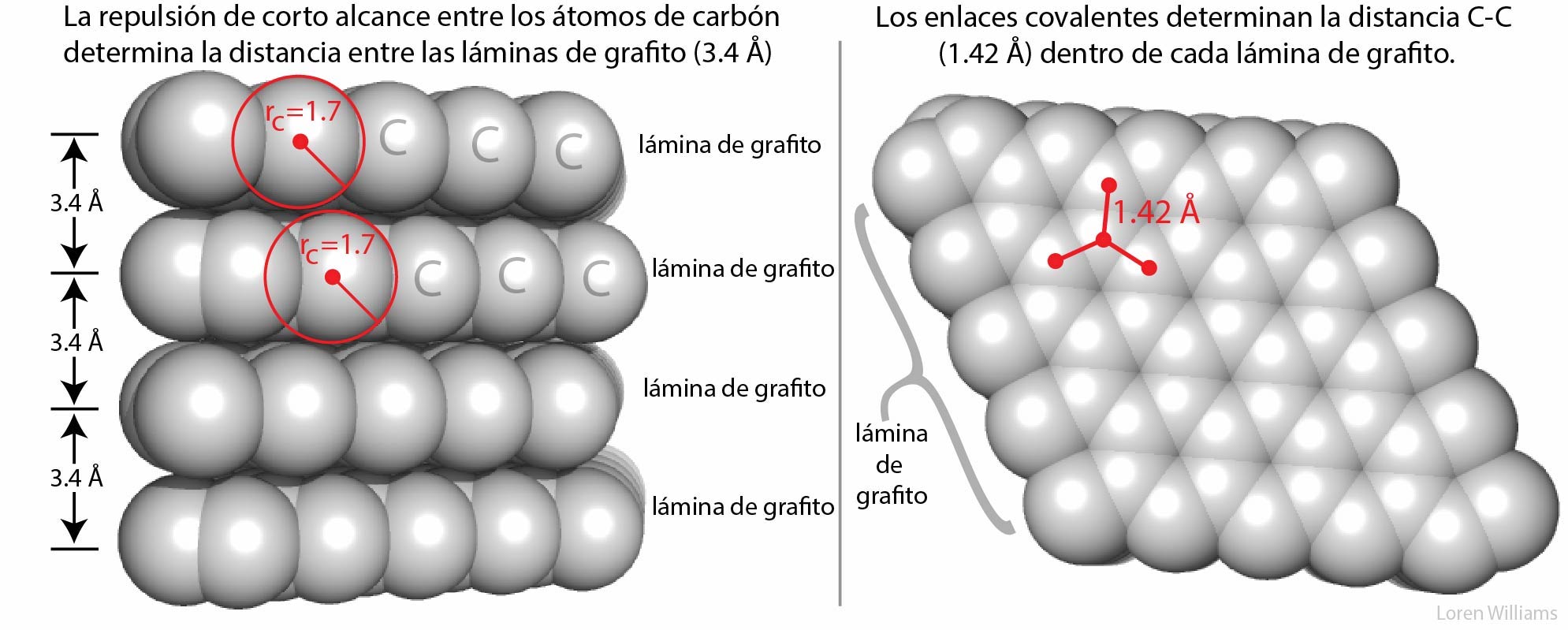
La figura 2 muestra cómo la repulsión de corto alcance establece la distancia de 3.4 Å entre láminas de grafito. Si dos átomos no enlazados se encuentran a una distancia menor que la suma de sus radios de van der Waals, las fuerzas de repulsión de corto alcance los mantienen separados.
La repulsión de corto alcance es importante en el día a día. Evita que las manos se crucen al aplaudir, y evita que los átomos colapsen en estados apretados de enorme densidad, del orden de 1014 g/ml, que es la densidad de los núcleos atómicos condensados. La gravedad muy alta, como en las estrellas de neutrones, sobrepasa la repulsión de corto alcance y hace que los átomos colapsen.
Aquí en la Tierra, con nuestra modesta gravedad, el radio de van der Waals del carbono (rC) resulta evidente al observar el espacio entre las capas de grafito. La distancia entre átomos en diferentes capas de grafito nunca es inferior al doble del radio de van der Waals del carbono (2 x rC = 2 x 1.7 = 3.4 Å). Los átomos dentro de una capa de grafito están conectados covalentemente (enlazados), lo que provoca la interpenetración de las superficies de van der Waals. Los átomos de carbono dentro de una capa están separados por 1.42 Å, que es mucho menos del doble del radio de carbono de van der Waals. Como se explica en otras secciones de este documento, las superficies de van der Waals también se violan cuando las moléculas forman enlaces de hidrógeno. Las coordenadas de grafito están aquí.
¿Cómo se siente la repulsión de corto alcance? Como intentar comprimir un líquido.
Las interacciones electrostáticas tienen lugar entre cationes y aniones, especies con carga formal de... -2, -1, +1, +2... Las interacciones electrostáticas pueden ser atractivas o repulsivas, dependiendo de los signos de las cargas. Las cargas del mismo signo se repelen. Las cargas de signo contrario se atraen.
Las interacciones electrostáticas favorables hacen que la presión de vapor del cloruro de sodio (y la de otras sales) sea muy baja. Si dejas cristales de sal de mesa (NaCl; Na+ = catión, Cl- = anión) en una sartén caliente, ¿cuánto tiempo pasa antes de que se vaporicen y sublimen? Un tiempo muy largo; las interacciones electrostáticas son extremamente fuertes. Las interacciones electrostáticas dentro de un cristal de cloruro de sodio se denominan 'enlaces iónicos'. Sin embargo, cuando un solo catión y un solo anión están juntos, dentro de una proteína o dentro de un ARN o ADN plegado, esas interacciones se consideran interacciones electrostáticas no covalentes. Las interacciones electrostáticas no covalentes pueden ser fuertes y actuar a largo alcance. Las fuerzas electrostáticas disminuyen gradualmente con la distancia (1/r2, donde r es la distancia entre los iones).

La figura 3 muestra la sección transversal de un cristal de NaCl. Cada catión de sodio experimenta fuertes interacciones electrostáticas con los aniones de cloro adyacentes. Las coordenadas del cloruro de sodio se pueden encontrar aquí.
Las interacciones electrostáticas son la principal interacción estabilizadora entre los oxígenos de los fosfatos del ARN (carga = -1) y los iones de magnesio (carga = +2), como se muestra en la siguiente figura. Hay muchos iones de magnesio asociados a ARN y ADN in vivo. Como se explica más adelante en este documento, las interacciones electrostáticas están altamente atenuadas (amortiguadas) por agua. En el plegamiento de proteínas, el plegamiento de ARN y la hibridación de ADN, las interacciones electrostáticas dependen de la concentración de sal y del pH en el entorno.
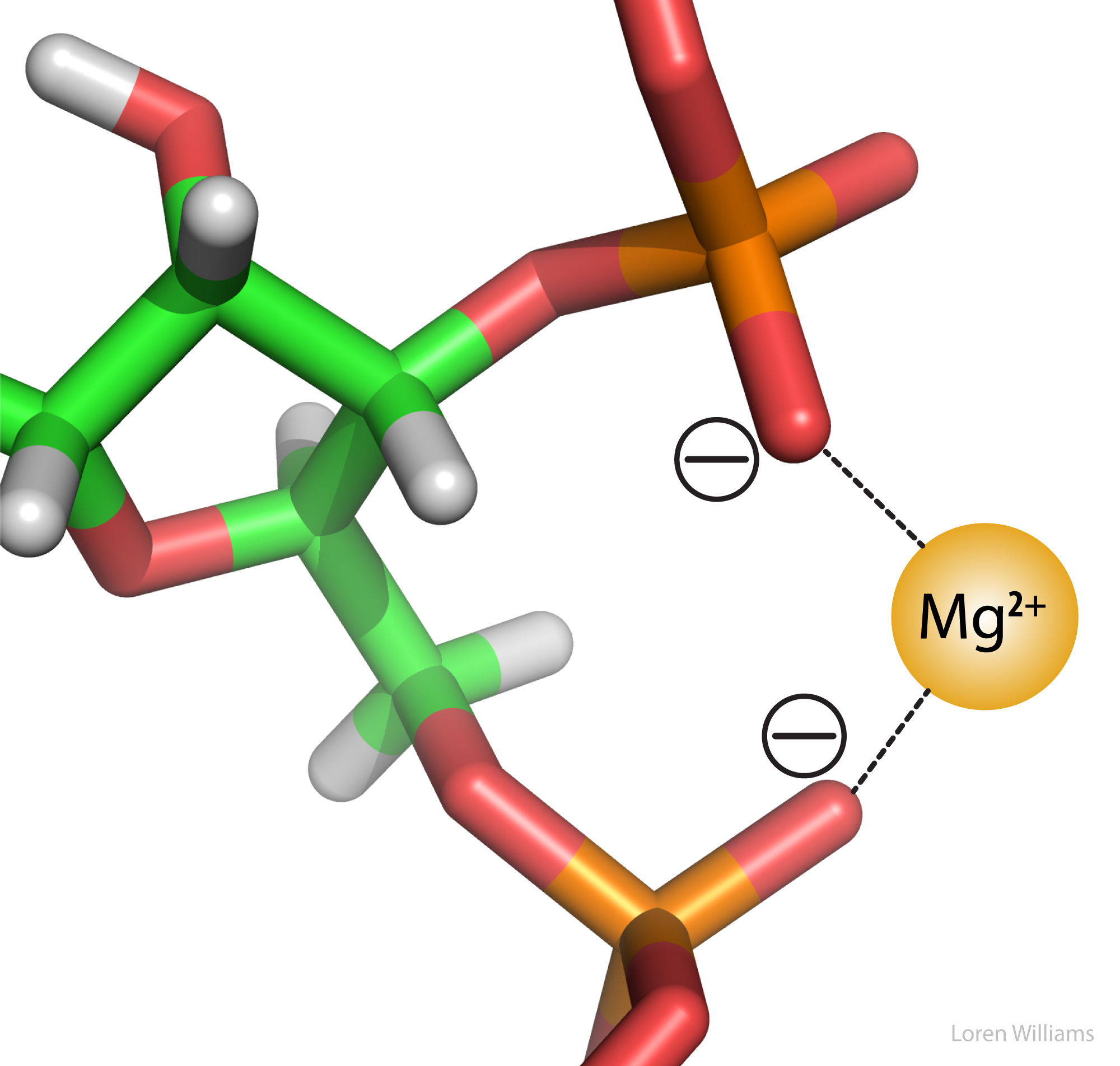
La figura 4 muestra interacciones electrostáticas. En el ARN (por ejemplo en el ribosoma), los átomos de oxígeno aniónicos de los fosfatos (carga formal = -1) establecen interacciones electrostáticas de atracción con los iones catiónicos de magnesio (carga +2). Dos grupos fosfato pueden “anclar” un átomo de Mg2+. La distancia entre los átomos de oxígeno y el Mg2+ es de 2.1 Å. Las líneas discontinuas representan interacciones electrostáticas favorables.
Las interacciones electrostáticas entre cadenas laterales de aminoácidos aniónicos y catiónicos son razonablemente frecuentes en las proteínas. Los pares de iones, a veces llamados 'puentes salinos', se forman cuando el grupo cargado de un aminoácido catiónico (como la lisina o la arginina) se sitúa alrededor de 3.0 a 5.0 Å del grupo cargado de un aminoácido aniónico (como el aspartato o el glutamato). Los grupos cargados en un par de iones generalmente están unidos por enlaces de hidrógeno, además de por interacciones electrostáticas.
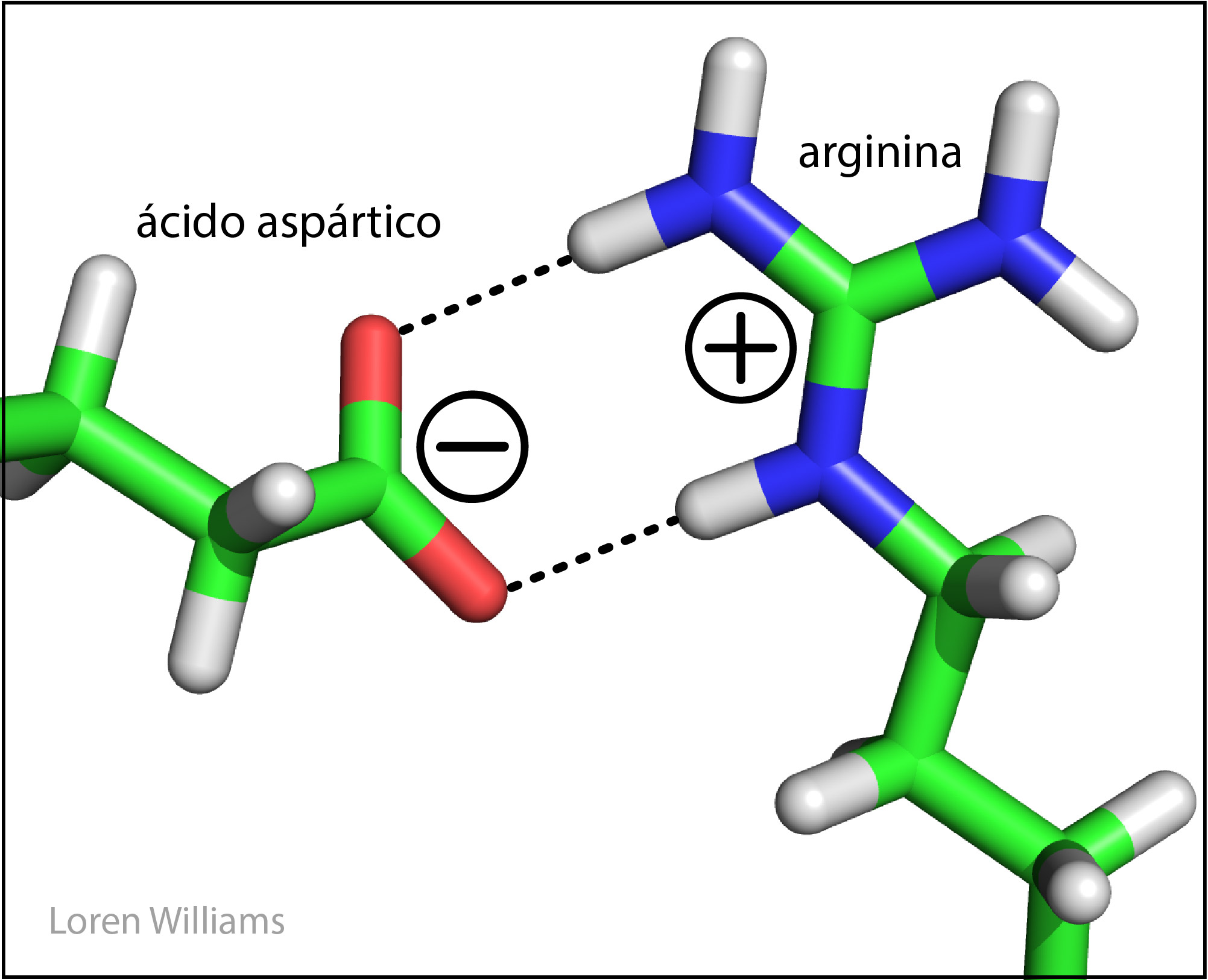
La figura 5 muestra un par iónico en una proteina plegada. Un ácido aspártico aniónico (carga = -1) participa en una interacción electrostática atractiva con una arginina catiónica (carga = +1). Las líneas discontinuas representan enlaces de hidrógeno.
La fuerza electrostática entre dos cargas puntuales viene dada por:
Fuerza = k q1 q2 / ε r2
donde k = 9.0 x 109 Nm2 / C2
q = -1.6 x 10-19 culombios para un electrón
r = distancia entre las cargas puntuales (metros)
ε = la constante dieléctrica del medio (adimensional).
ε es la constante dieléctrica. ε refleja la tendencia del medio a apantallar especies cargadas unas de las otras. ε es 1 en el vacío, alrededor de 4 en el interior de una proteína y 80 en el agua. Calcular los efectos electrostáticos en sistemas biológicos es complejo, en parte debido a la falta de uniformidad del entorno dieléctrico. Los microambientes dieléctricos son complejos y variables, con menor apantallamiento de cargas en regiones de cadenas laterales de hidrocarburos y mayor apantallamiento en regiones de cadenas laterales polares.
La energía electrostática viene dada por:
ΔE= k a q1 q2 / ε r
donde a = número de Avogadro.
Se puede estimar aproximadamente la energía de interacción carga-carga en una proteína. La energía de una amina (carga +1) y un ácido carboxílico (carga -1) separadas 4 Å en el interior de una proteína viene dada por:
ΔE = -(9.0x109nt-m2/C2)(6.02x1023)(1.6x10-19C)2 /4( 4x10-10m)
= 87 kJ/mol = 21 kcal/mol
Este cálculo aproximado es unas 10 veces mayor que los valores determinados experimentalmente. Un par iónico contribuye con un ΔG favorable de 1 a 4 kcal/mol (4.1 a 16.4 kJ / mol) a la estabilidad de una proteína nativa.
Nota sobre la nomenclatura.
Las fuerzas de atracción entre un ion Mg2+ y grupos fosfato (arriba) se denominan interacciones electrostáticas. Las especies con carga formal (2, 1, -1, -2) participan en interacciones electrostáticas. Utilizamos otros términos (dipolo-dipolo...) para describir las interacciones entre cargas parciales. El conjunto de nombres es confuso porque TODAS las interacciones moleculares son entre electrones y electrones, y entre electrones y núcleos, y de echo son de naturaleza electrostática. Podría haber sido mejor usar nombres diferentes que tengan más sentido. Sin embargo, por convención tenemos que restringir el término 'electrostático' a las interacciones entre especies formalmente cargadas.
Electronegatividades. Para poder comprender las interacciones dipolares, es necesario conocer la electronegatividad. Los electrones no se reparten por igual en una molécula con átomos diferentes. La tendencia de cualquier átomo a atraer electrones hacia sí mismo, y lejos de otros átomos, se caracteriza por una magnitud llamada electronegatividad. El flúor es el átomo más electronegativo (4.0) y el cesio el menos electronegativo (0.7). En general, la electronegatividad aumenta con la carga nuclear mientras se mantiene constante el número de electrones centrales (es decir, de izquierda a derecha en una fila de la tabla periódica). La electronegatividad aumenta a medida que disminuye el apantallamiento nuclear (de abajo a arriba en una columna de la tabla periódica).
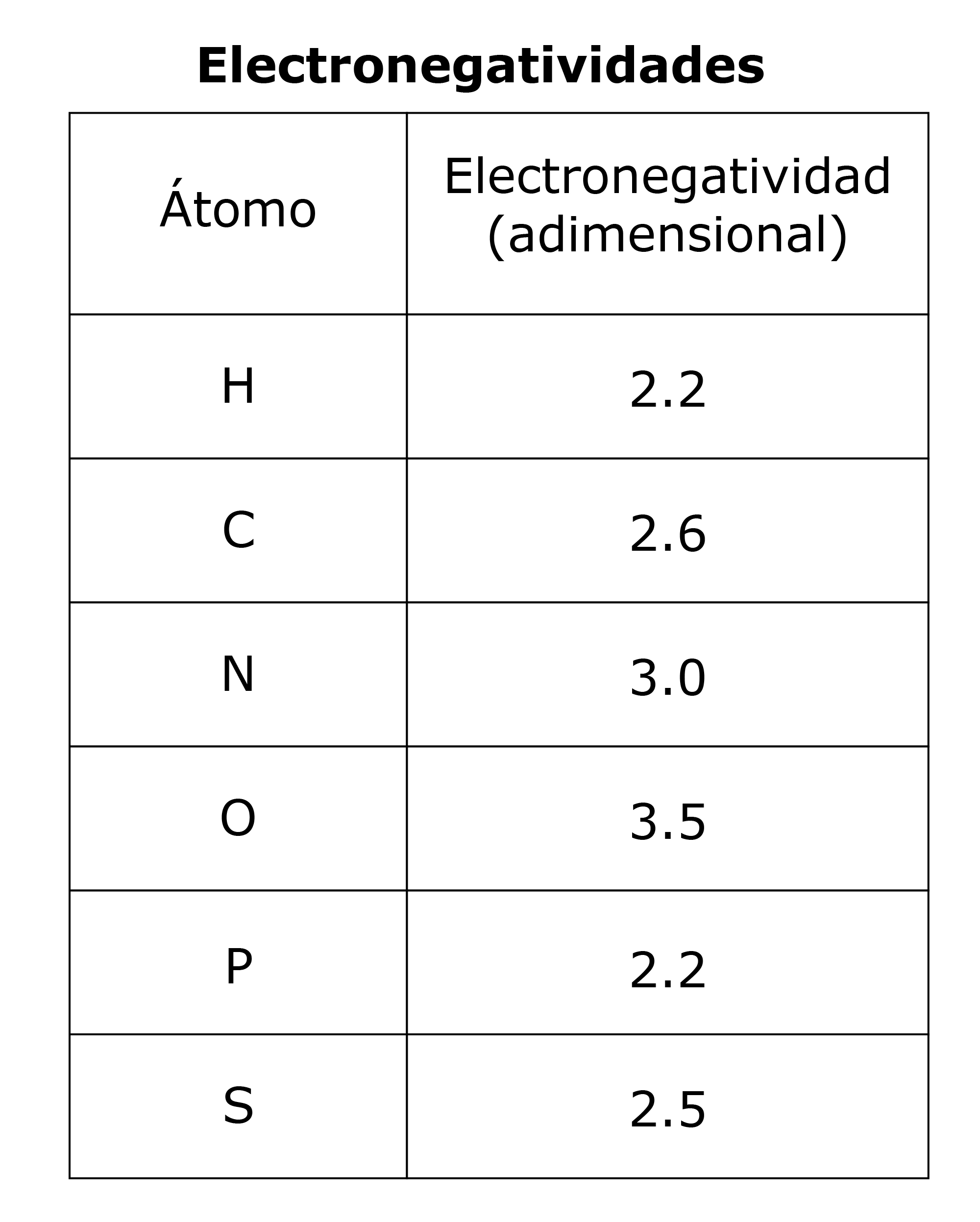
Cargas parciales. En una molécula compuesta por átomos de diversas electronegatividades, los átomos con electronegatividades más bajas (de magnitud más pequeña) tienen cargas parciales positivas (δ+) y los átomos con las electronegatividades más elevadas tienen cargas parciales negativas (δ-). Una mayor diferencia en las electronegatividades de dos átomos enlazados hace que el enlace entre ellos sea más polar y que las cargas parciales en los átomos sean de mayor magnitud. En los sistemas biológicos, el oxígeno es generalmente el átomo más electronegativo y tiene la mayor carga parcial negativa.
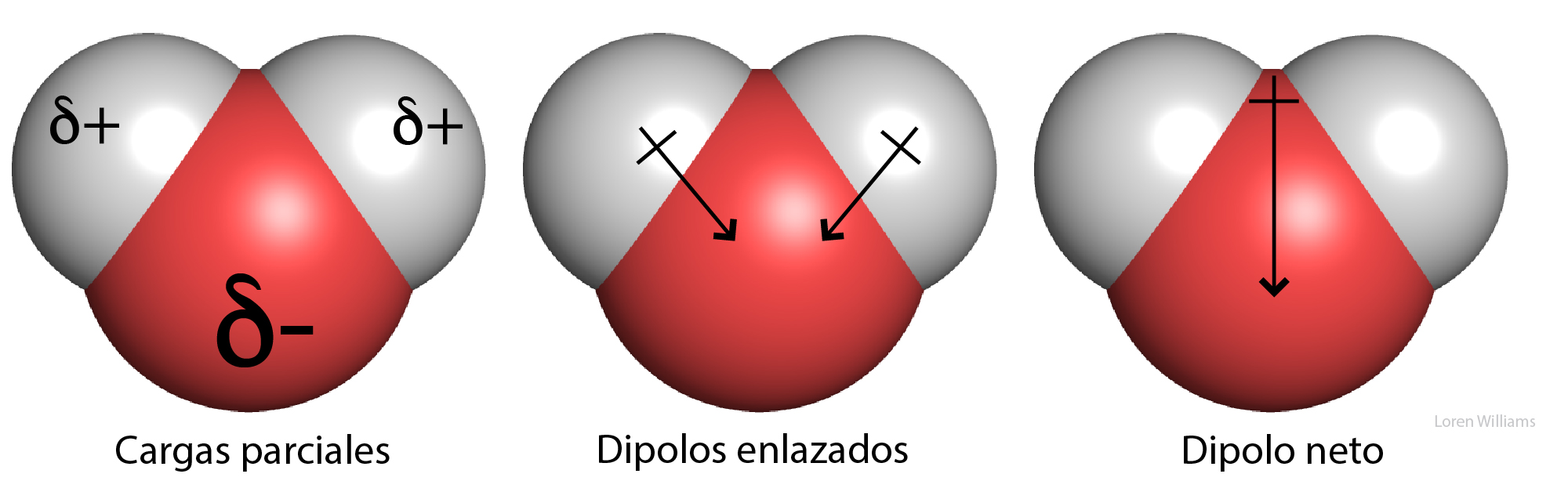
La figura 6 muestra la distribución de carga y momento dipolar de la molécula de agua. El átomo electronegativo de oxígeno atrae la densidad electronegativa de los átomos de hidrógeno. El oxígeno es portador de una carga parcial negativa y los átomos de hidrógeno contienen carga parcial positiva. Los dipolos enlazados (centro) y los dipolos moleculares (derecha) pueden ser representados como un vector. Las flechas indican la dirección de carga positiva a carga negativa.
En el metanol (CH3OH), el átomo electronegativo de oxígeno atrae la densidad electrónica de los átomos de carbono e hidrógeno. En el agua (H2O), el átomo electronegativo de oxígeno atrae la densidad electrónica de ambos átomos de hidrógeno. El átomo de oxígeno del agua tiene una carga negativa parcial. Los átomos de hidrógeno tienen cargas positivas parciales. Este fenómeno de separación de carga se llama polaridad. El metanol y el agua son moléculas polares. El nitrógeno molecular (N2) es una molécula no polar porque los dos átomos de nitrógeno tienen electronegatividades iguales y, por lo tanto, comparten los electrones por igual. Los hidrocarburos (CH3CH2... CH2CH3) no son polares porque las electronegatividades de carbono e hidrógeno son similares.
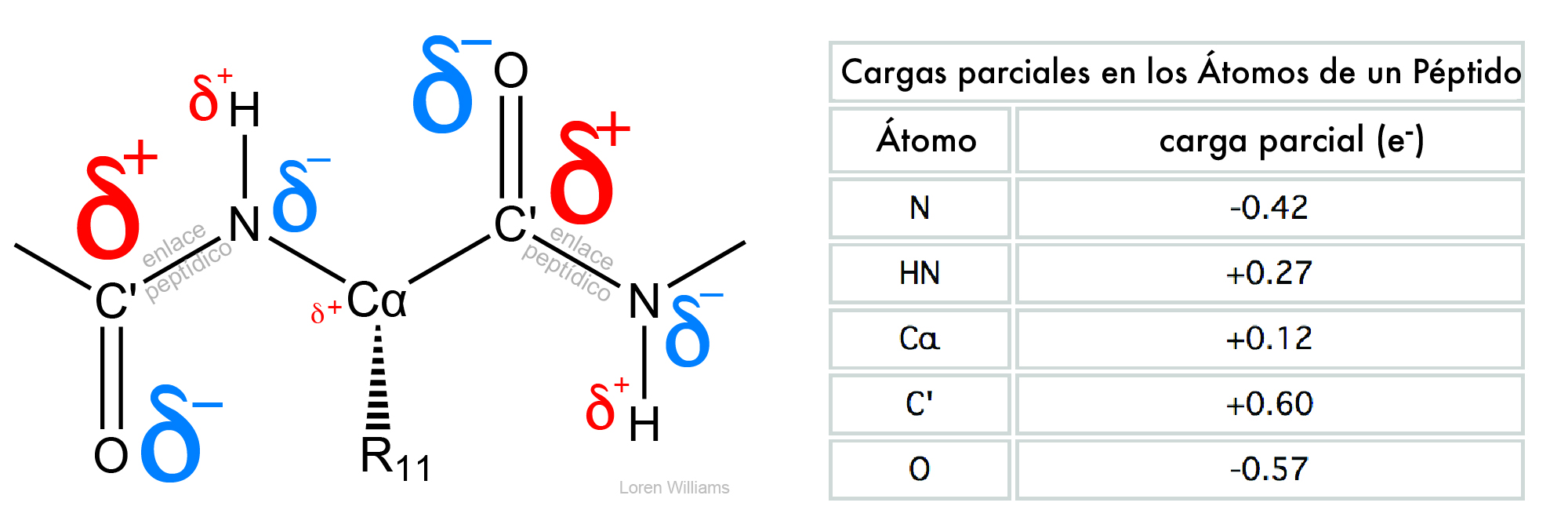
La figura 7 muestra las cargas parciales dentro de un polipéptido. El tamaño del símbolo (δ) es proporcional a la magnitud de la carga parcial. Los átomos de oxígeno son los más electronegativos y en consecuencia tienen la carga parcial negativa más grande.
Momento dipolar. El grado de separación de la carga dentro de una molécula se caracteriza por el momento dipolar (μ). El momento dipolar está determinado por las magnitudes de las cargas parciales y por la distancia entre ellas. Para cuantificar los momentos dipolares, las cargas se expresan en esu y las distancias en centímetros. El momento dipolar de un electrón y un protón separados por 1 Å es igual a:
(4.8 x 10-10 esu) (10-8cm) = 4.8 x 10-18 esu cm
= 4.8 Debye
El momento dipolar del agua es 1.85 Debye (HCl = 1.1 D; CH3Cl = 1.9 D; HCN = 2.9 D; NH3 = 1.47).
La orientación del momento dipolar de un péptido es aproximadamente paralela al enlace N-H y es de alrededor de 3.7 Debye en magnitud.
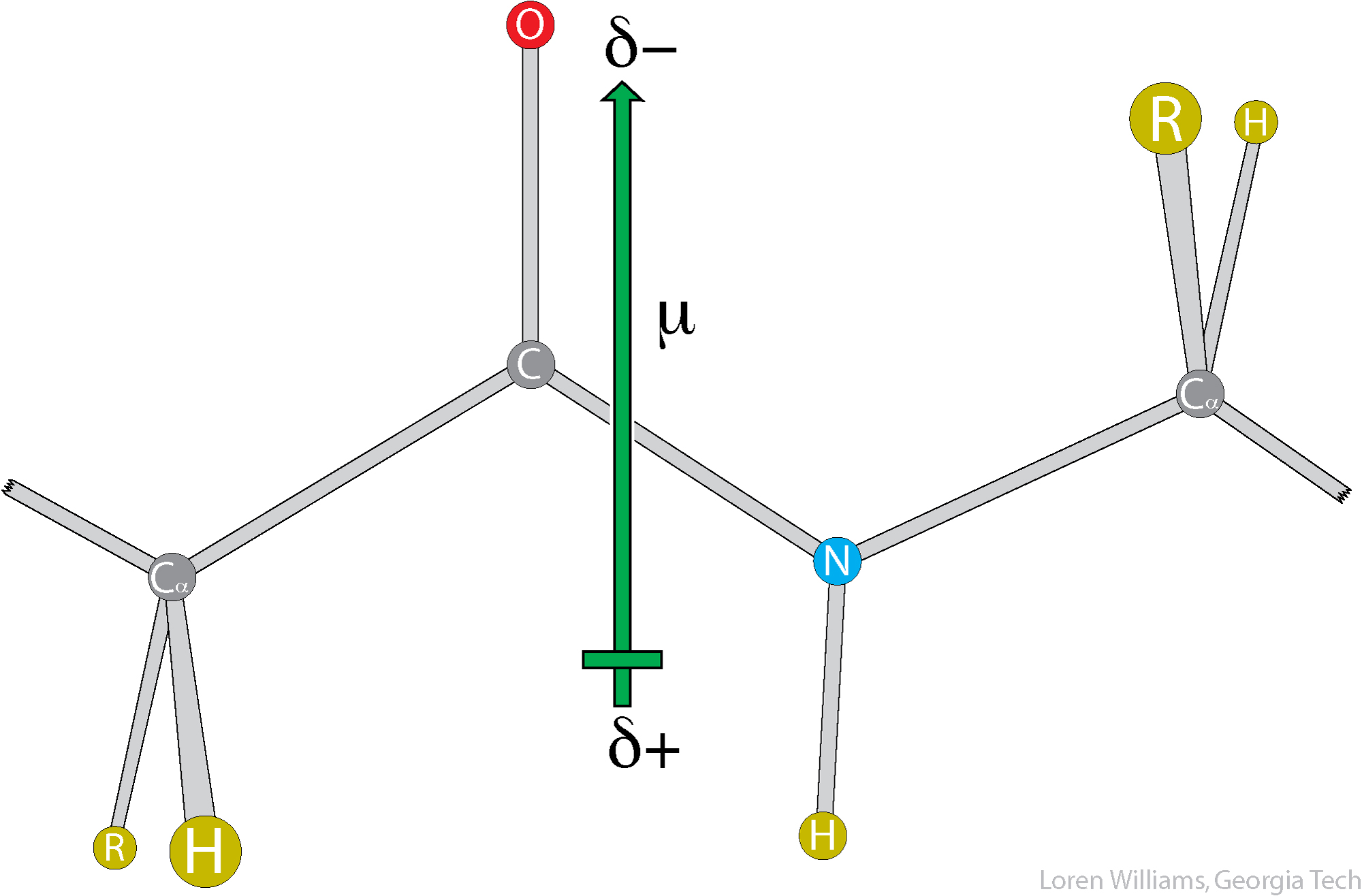
La figura 8 muestra la orientación del momento dipolar en un péptido.
El elevado momento dipolar de un enlace peptídico sugiere que las interacciones dipolares son importantes para la conformación e interacciones de las proteínas. Y, en efecto, lo son.
Un dipolo está rodeado por un campo eléctrico, lo que genera fuerzas a distancia sobre las especies cargadas y parcialmente cargadas en sus inmediaciones. Las interacciones entre dipolos e iones se denominan interacciones carga-dipolo (o interacciones ión-dipolo). Los dipolos también interactúan con otros dipolos (interacciones dipolo-dipolo), e inducen la redistribución de cargas (polarización) en las moléculas circundantes (interacciones dipolo-dipolo inducido). En las próximas secciones discutiremos por separado cada una de estas interacciones.
Dos dipolos se sienten entre sí a cierta distancia. El extremo positivo del primer dipolo es atraído hacia el extremo negativo del segundo y es repelido por el extremo positivo. La fuerza de una interacción dipolo-dipolo depende del tamaño de ambos dipolos, de su proximidad y de sus orientaciones relativas. La energía de interacción neta entre dos dipolos puede ser positiva o negativa. Los dipolos paralelos se atraen mientras que los anti-paralelos se repelen. En la siguiente figura podemos ver las energías de interacción para varias orientaciones de dos dipolos con momentos de 1 Debye a una distancia de 5 Å en un entorno de ε = 4.
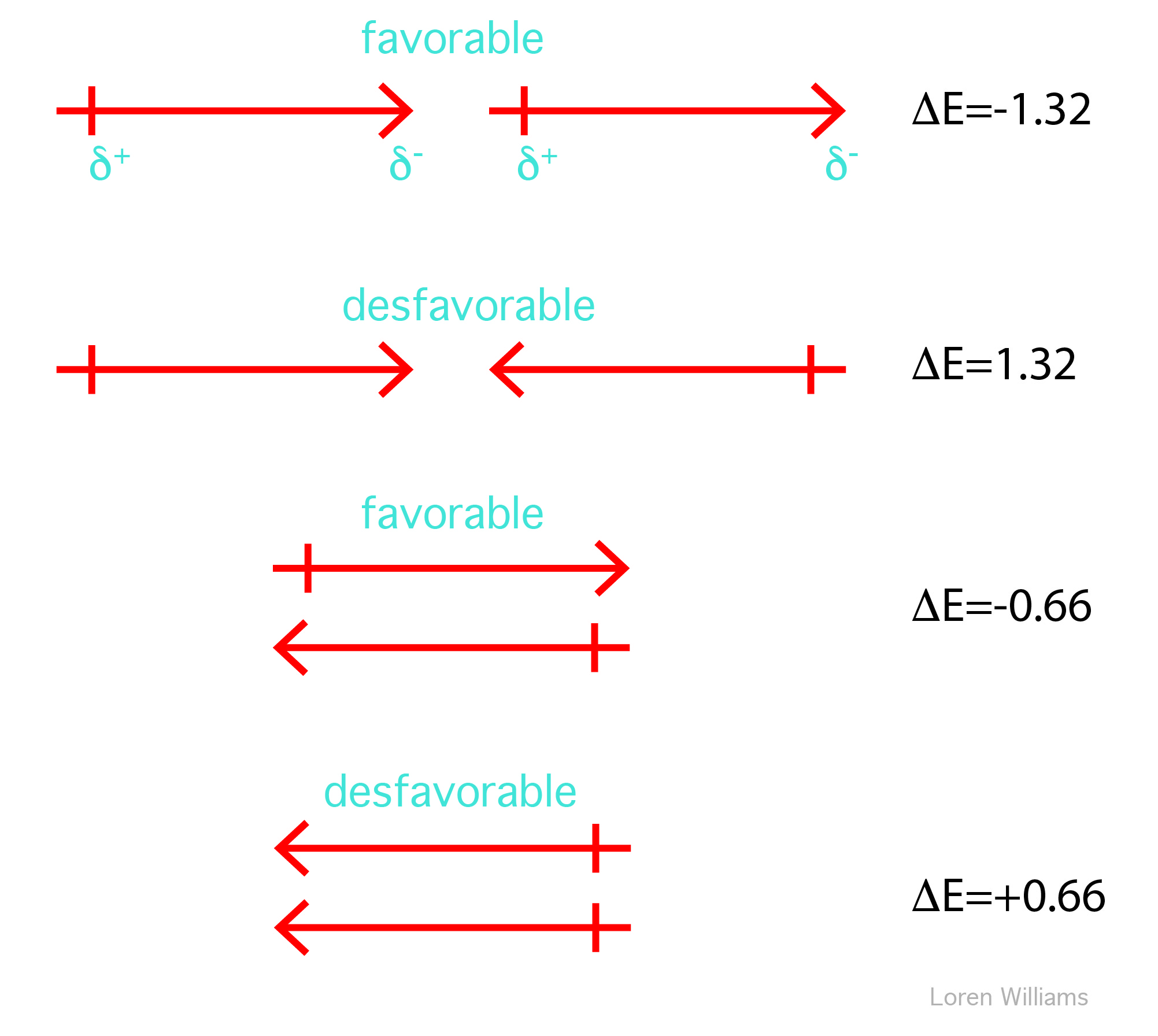
La figura 9 muestra las interacciones dipolo-dipolo dependen de las orientaciones de los dipolos. Las interacciones dipolo-dipolo pueden ser atractivas o repulsivas.
En los fluidos, la orientación de los dipolos moleculares cambia rápidamente a medida que las moléculas cambian de posición. Sin embargo, sus momentos dipolares tienden a orientarse favorablemente. Por consiguiente, en acetona líquida, por ejemplo, las interacciones dipolo-dipolo tiene mayor peso que las interacciones dipolo-dipolo desfavorables. Las interacciones dipolo-dipolo pierden importancia con 1/r3.
La densidad electrónica de una molécula polarizable se desplaza y deforma por los campos eléctricos de las moléculas polares circundantes.

Cualquier molécula con un momento dipolar (o cualquier ion) está rodeada por un campo electrostático. Este campo electrostático desplaza la densidad de electrones (altera los momentos dipolares) de las moléculas cercanas. Un cambio en el momento dipolar de una molécula debido a otra molécula (o a cualquier campo eléctrico externo) se llama polarización. La facilidad con la que la densidad electrónica se desplaza por un campo electrónico se llama polarizabilidad. Los átomos grandes como el xenón son más polarizables que los átomos pequeños como el helio. Las cadenas laterales de aminoácidos con electrones π, como la fenilalanina y el triptófano, son más polarizables que las cadenas laterales como la isoleucina, que carecen de electrones π.
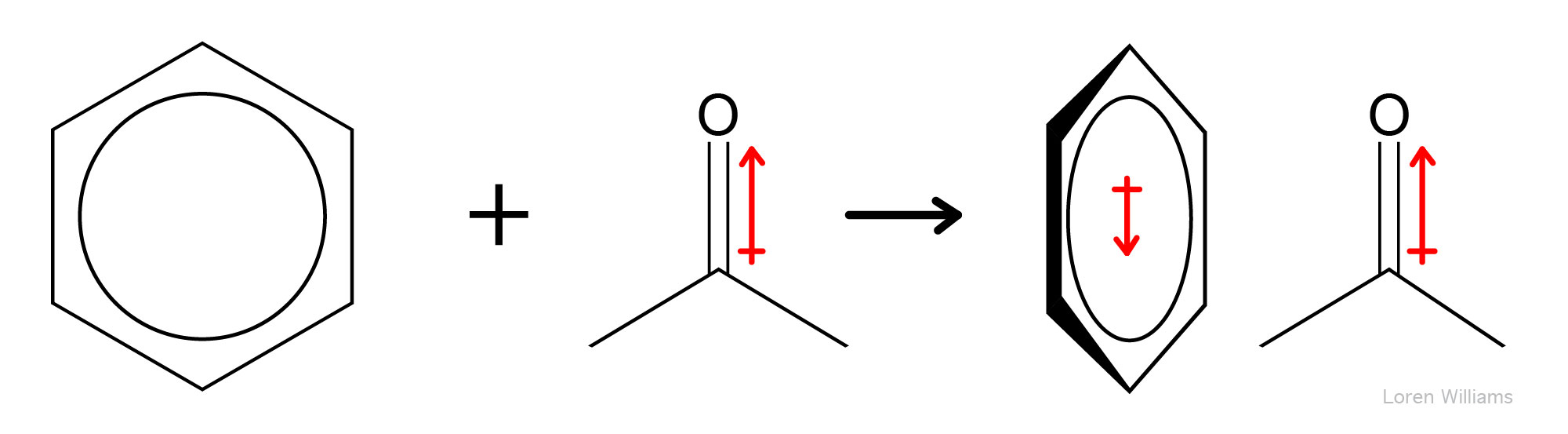
La figura 10 muestra cómo un dipolo estático puede inducir un dipolo en una molécula adyacente. Cuando dos moléculas aisladas (izquierda) se acercan en un líquido o sólido (derecha), el dipolo estático 'polariza' la molécula adyacente. Los electrones π son más polarizables (se perturban más fácilmente por un dipolo adyacente) que los electrones σ. La fuerza de una interacción dipolo inducida por dipolo depende del tamaño del momento dipolar de la primera molécula y de la polarizabilidad de la segunda molécula.
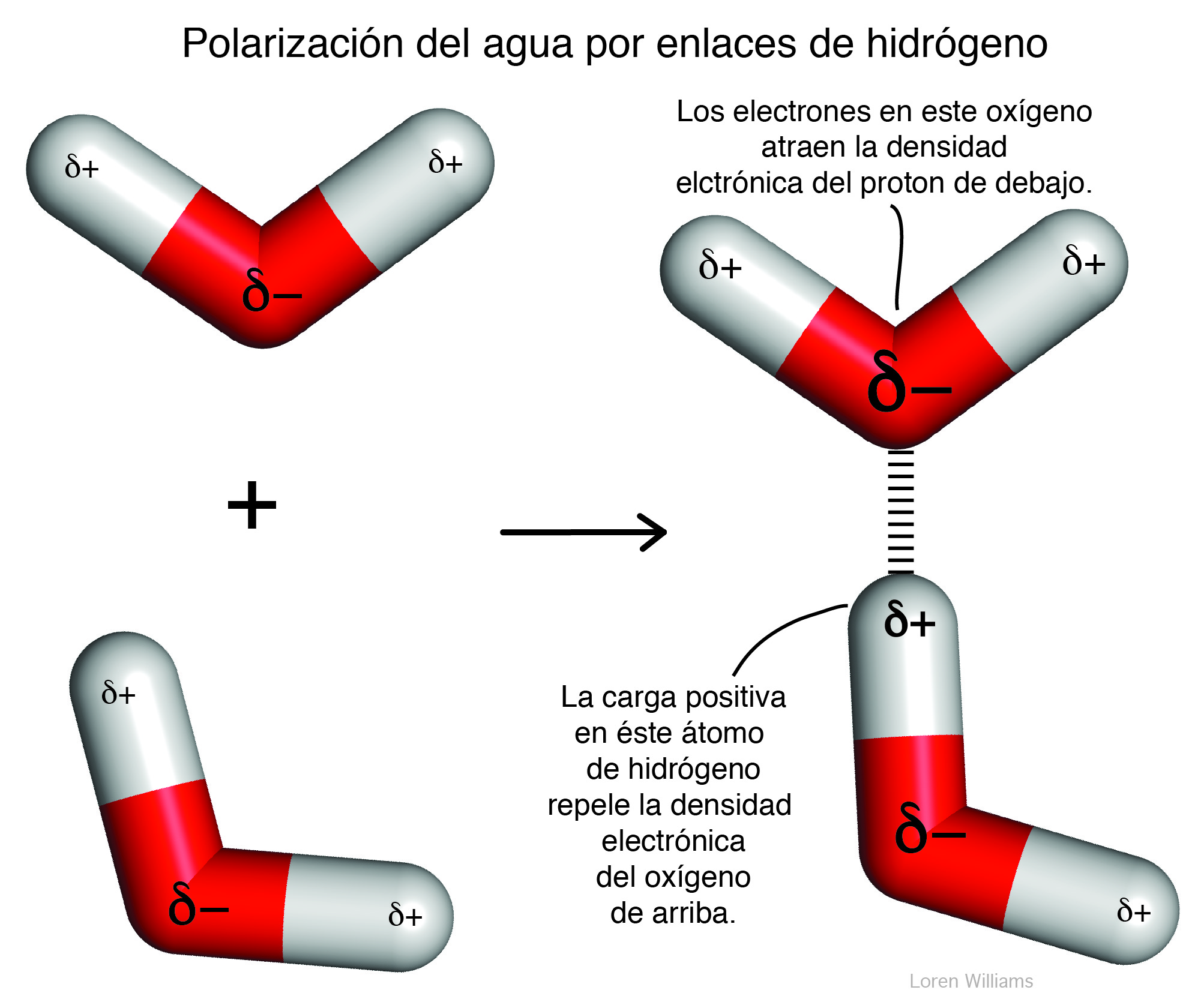
La figura 11 muestra cómo las moléculas de agua se polarizan entre sí. Cada molécula de agua polariza las moléculas de agua vecinas y aumenta los momentos dipolares vecinos. Cuando dos moléculas de agua se acercan entre sí y forman un enlace de hidrógeno como se muestra aquí, la carga negativa parcial en el oxígeno de la molécula de agua superior aumenta en magnitud, y la carga positiva parcial en el protón de la molécula de agua inferior también aumenta. El tamaño del símbolo es proporcional a la magnitud de la carga parcial.
Las interacciones dipolo-dipolo inducido son importantes incluso entre moléculas con dipolos permanentes. Un dipolo permanente es perturbado por un dipolo adyacente. Por ejemplo, en agua líquida (donde las moléculas están muy juntas), todas las moléculas de agua están polarizadas. El dipolo permanente de cada molécula de agua polariza todas las moléculas de agua adyacentes. El dipolo de una molécula de agua induce cambios en los dipolos de toda molécula de agua cercana.
Las interacciones dipolo-dipolo inducido son siempre atractivas y pueden contribuir hasta 0.5 kcal/mol (2.1 kJ / mol) a la estabilización de las asociaciones moleculares. Las interacciones dipolo-dipolo inducido decaen como 1/r4. Las especies formalmente cargadas (Na+, Mg2+, -COO-, etc.) también polarizan las moléculas cercanas e inducen dipolos favorables. Las interacciones resultantes reciben el nombre de interacciones de dipolo inducidas por carga (o interacciones de dipolo inducidas por iones). Estas interacciones son importantes, por ejemplo, en la estructura de las proteínas, aunque no les dediquemos una sección separada en este documento.
Una molécula con un dipolo permanente puede interactuar favorablemente con cationes y aniones. Este tipo de interacción se llama interacción carga-dipolo o ion-dipolo. Las interacciones carga-dipolo son la razón por la cual el cloruro de sodio (compuesto de iones catiónicos de sodio e iones aniónicos de cloruro) y otras sales tienden a interactuar bien con el agua, y son muy solubles en agua, que tiene un fuerte dipolo.
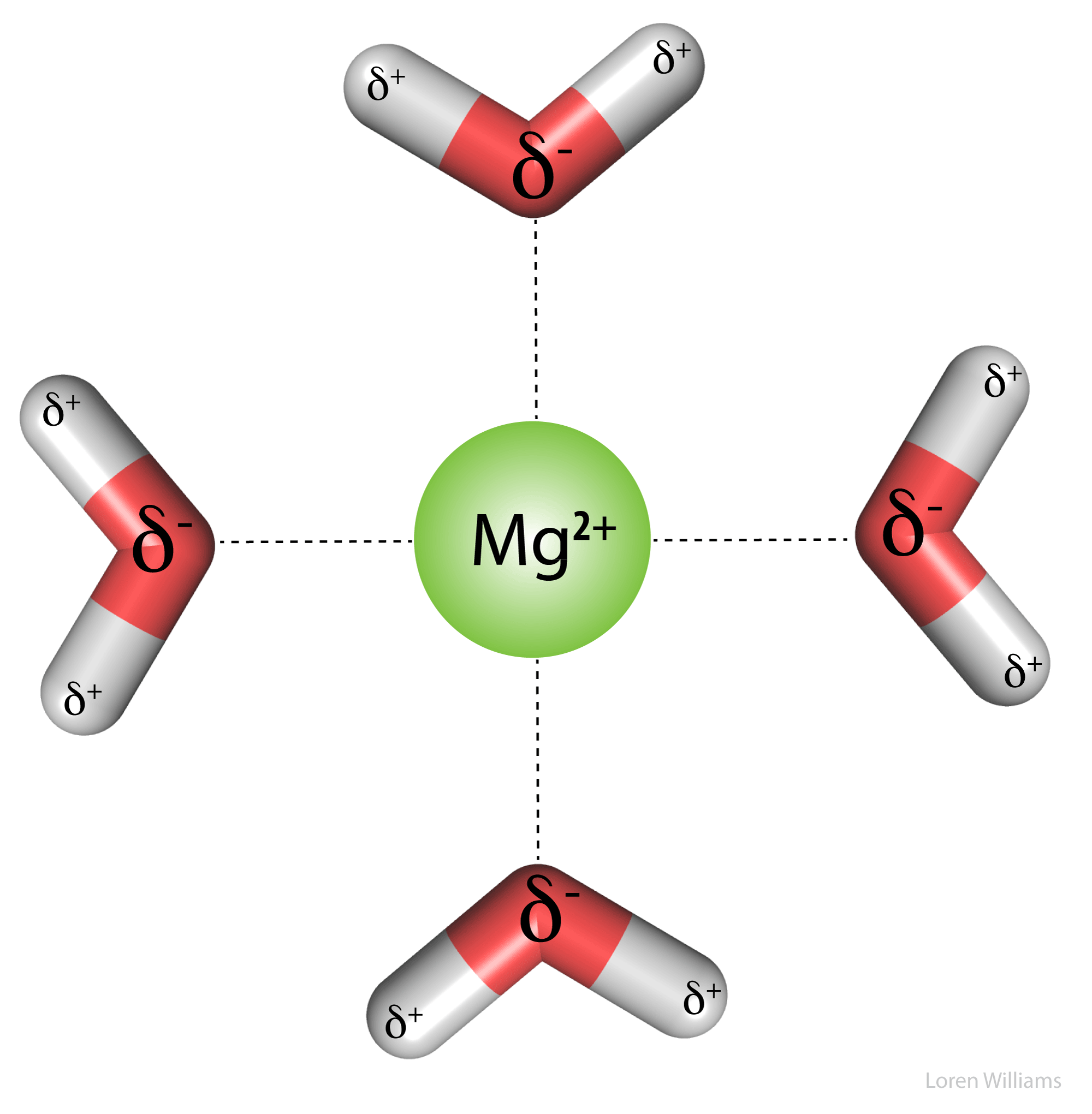
La figura 12 muestra las interacciones carga-dipolo. Cuatro moléculas de agua interactúan favorablemente con un catión divalente de magnesio. Los extremos negativos de los dipolos de agua se dirigen hacia el ion magnesio cargado positivamente. Seis moléculas de agua coordinan el magnesio en solución. Dos de ellos se han omitido para mayor claridad. En el caso de un anión como el cloruro, las moléculas de agua cambian de dirección y dirigen los extremos positivos de sus dipolos hacia el anión. Aquí las líneas discontinuas no representan enlaces de hidrógeno. No hay átomos de hidrógeno entre el catión Mg2+ y los átomos de oxígeno del agua.
Podemos ver resonancia a nuestro alrededor. Un niño en un columpio, las mareas o las cuerdas de un violín ilustran frecuencias de resonancia naturales de sistemas físicos. El Puente de Tacoma Narrows es uno de los ejemplos más famosos de resonancia.
Las moléculas también resuenan. Los electrones, incluso en un átomo esférico como el helio o el xenón, fluctúan con el tiempo de acuerdo con la frecuencia natural de resonancia de ese átomo. Aunque los químicos describan los átomos de helio o xenón como esféricos, si se pudiese tomar una imagen instantánea de uno de estos átomos, siempre se encontraría en un estado transitorio no-esférico. El xenón es esférico en promedio, pero no en un punto temporal instantáneo.
Al fluctuar la densidad electrónica, los momentos dipolares también fluctúan. Por tanto, todas las moléculas y los átomos contienen dipolos oscilantes. En todas las moléculas que se encuentran situadas en proximidad (en cualquier líquido o solido, pero no en un gas perfecto), los dipolos oscilantes interactúan entre si y se acoplan. Oscilan en sincronía, como las cuerdas de un violín. Los movimientos de los electrones en moléculas adyacentes están correlacionados. Los electrones en una molécula tienden a desplazar los de las moléculas cercanas, debido a la repulsión electrostática. Los dipolos fluctuantes acoplados experimentan interacciones electrostáticas favorables conocidas como interacciones dispersivas.
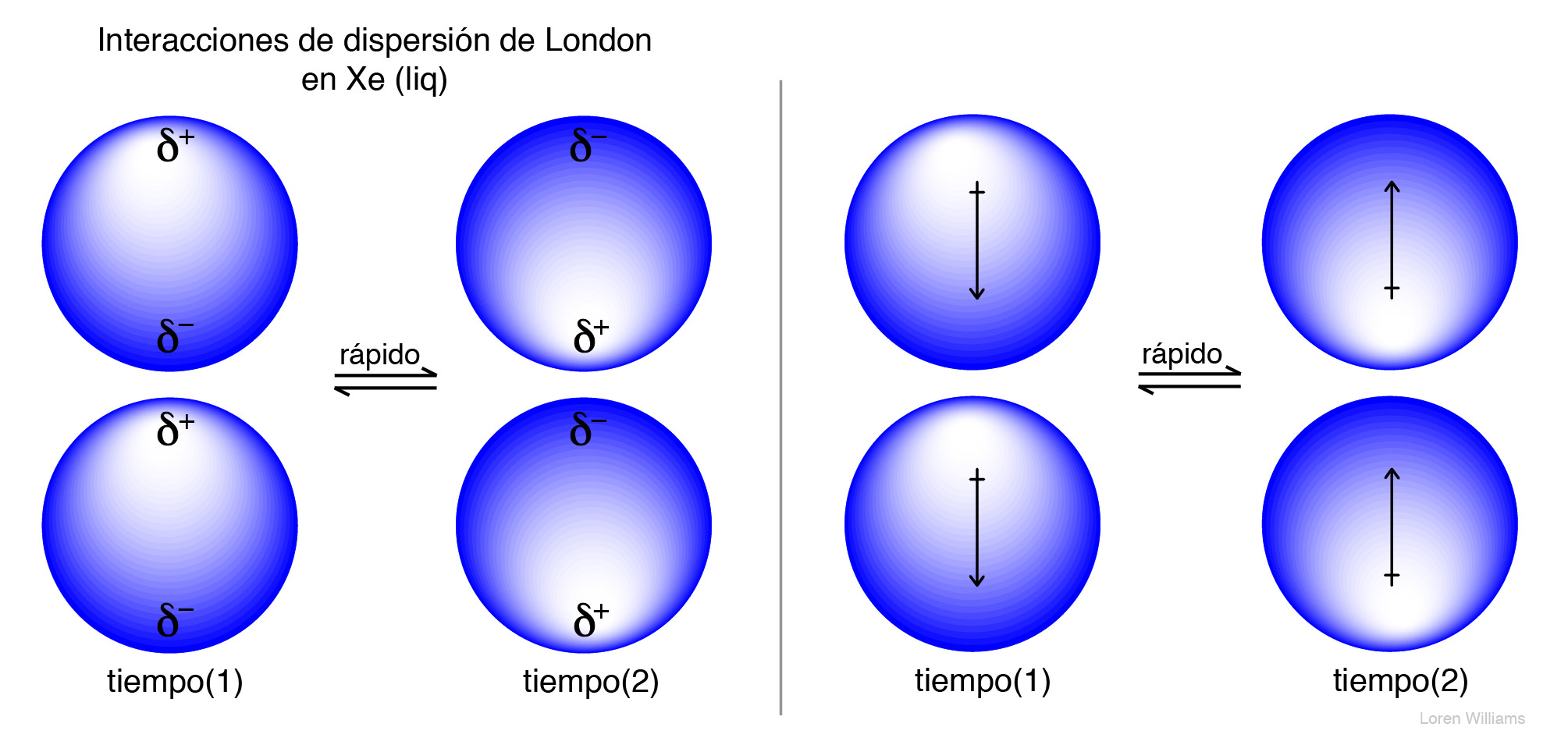
La figura 13 muestra cómo las interacciones dispersivas en Xenón líquido (o Helio o Neón, etc.) son causadas por interacciones atractivas entre fluctuaciones acopladas de sus dipolos. La zona azul más oscura indica una mayor densidad de electrones. Las fluctuaciones están correlacionadas y son muy rápidas, en la escala de tiempo de femtosegundos (10-15 segundos). Los átomos de xenón adyacentes experimentan la atracción electrostática de los dipolos transitorios. Aquí se muestran dos representaciones diferentes de dipolos fluctuantes.
Las interacciones dispersivas son siempre atractivas y ocurren entre cualquier par de moléculas cercanas, polares o no polares. Las interacciones dispersivas son las únicas fuerzas atractivas entre átomos en helio líquido He (punto de ebullición 4.5 K), neón Ne (27 K), argón Ar (87 K) y entre moléculas de nitrógeno N2 (77 K). Sin interacciones dispersivas no habría estado líquido para los gases nobles.
El número total de pares de interacciones dispersivas átomo-átomo en una proteína plegada es enorme, de manera que las interacciones dispersivas pueden contribuir sustancialmente a la estabilidad. La fuerza de esta interacción está relacionada con la polarizabilidad. Los aminoácidos triptófano, tirosina, fenilalanina e histidina son los que tienen las cadenas laterales mas polarizables, y forman las interacciones dispersivas más fuertes en las proteínas.
¿Qué hay del agua? Incluso moléculas con dipolos permanentes, como el agua, experimentan interacciones dispersivas. En torno a 25% de las fuerzas atractivas entre las moléculas de agua en fase líquida son de naturaleza dispersiva.
Las interacciones dispersivas decaen como 1/r6.
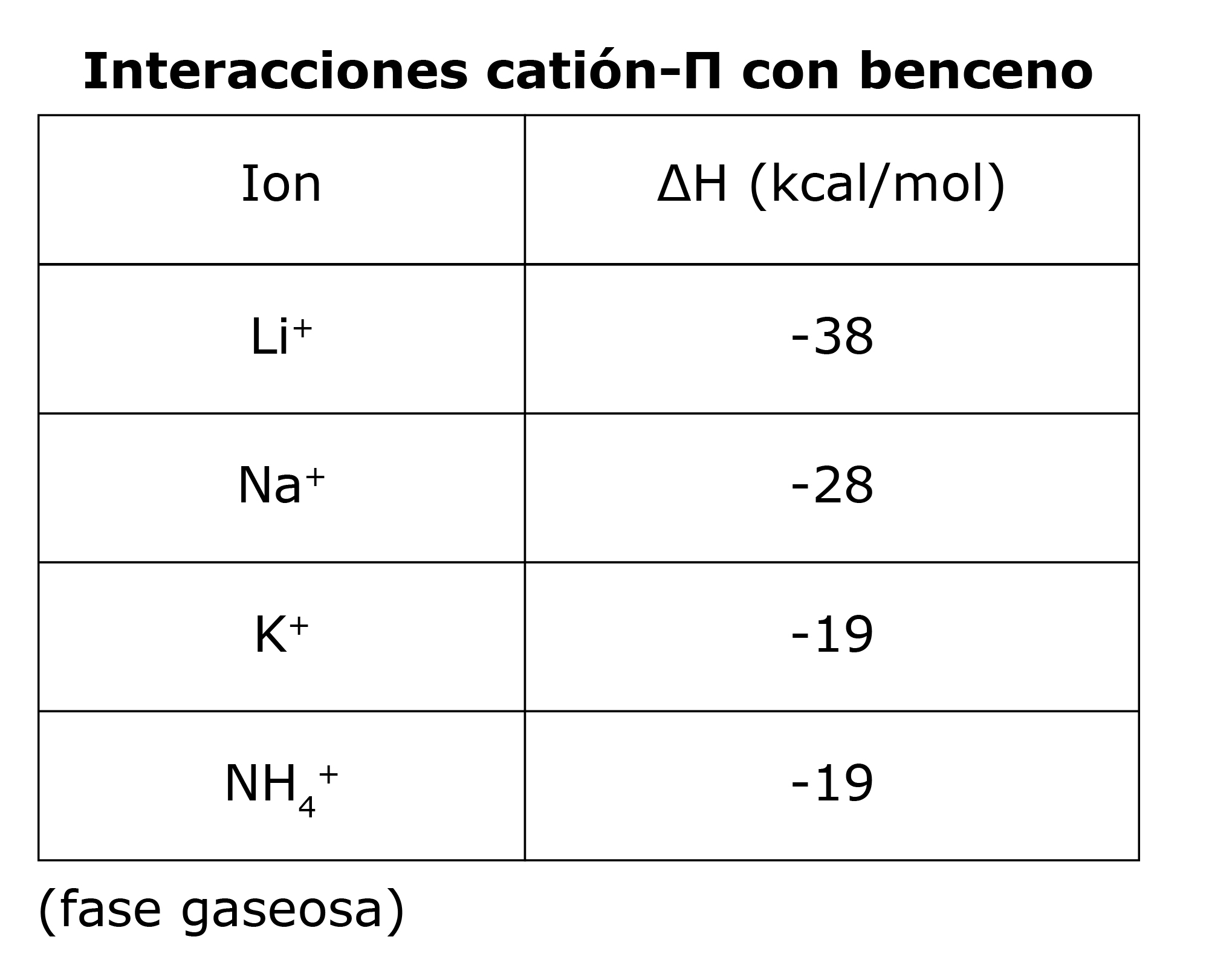
Un sistema Π como el benceno, el triptófano, la fenilalanina o la tirosina concentra carga negativa parcial por encima y por debajo del plano del anillo. Un catión puede interaccionar favorablemente con esta carga negativa cuando está cerca de la cara del sistema Π. En los complejos más estables de este tipo, el catión es centrado directamente sobre el sistema Π y se encuentra en contacto directo con él vía fuerzas de van der Waals. La tabla de la izquierda muestra entalpías de interacción en fase gas de varios cationes, que son del mismo orden que sus entalpías de hidratación. Por lo tanto, las interacciones catión-Π son de fuerza más o menos similar a la de las interacciones catión-dipolo formadas entre el agua y el catión. Los iones pequeños con alta densidad de carga forman complejos catión-Π más fuertes que los iones grandes. Los grupos electrófilos en el sistema del anillo debilitan las interacciones con el catión mientras que los grupos que donan electrones la fortalecen.
Las interacciones catión-Π son importantes para la estructura de las proteínas. El grupo guanidino de la arginina y el ε-NH3+ de la lisina establecen interacciones catión-Π con cadenas laterales aromáticas de las proteínas. Un par catión-Π en condiciones favorables contribuye tanto a la estabilidad proteica como un enlace de hidrógeno o una interacción electrostática (carga-carga). El sistema Π más habitual en proteínas es el triptófano mientras que el catión más común es la arginina. Triptófano y arginina pueden formar estructuras coplanares grandes.
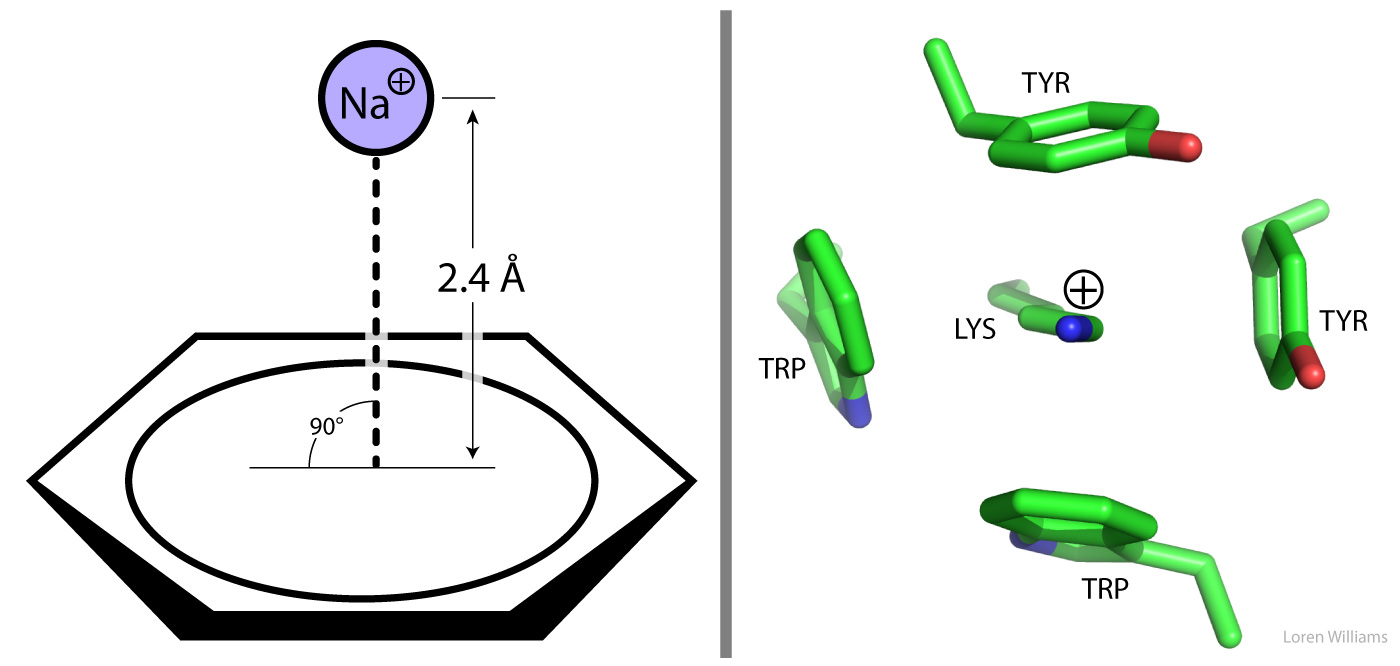
La figura 14 (izquierda) muestra la geometría óptima para una interacción catión-Π entre un catión Na+ y benceno. La distancia desde el ión Na+ al centro del anillo es 2.4 Å (radio iónico de Na+ = 0.9 Å, radio de van der Waals del carbono (rC) = 1.7 Å). (Derecha) El ε-NH3+ de una lisina participa en interacciones de catión-Π con dos cadenas laterales de triptófano y dos cadenas laterales de tirosina en una proteína (glucoamilasa, PDB ID 1GAI).
La idea de que un solo átomo de hidrógeno pueda enlazarse simultáneamente con otros dos átomos fue propuesta en 1920 por Latimer y Rodebush y su director de tesis G.N. Lewis. Maurice Huggins, que también fue un estudiante en el grupo de Lewis, describió los enlaces de hidrógeno en su tesis de 1919.
Un enlace de hidrógeno es una interacción favorable entre un átomo con un par de electrones solitarios (una base de Lewis) y un átomo de hidrógeno que ha perdido una parte de sus electrones porque está enlazado covalentemente con un átomo electronegativo (N, O o S). En un enlace de hidrógeno, la base de Lewis es el aceptor del enlace de hidrógeno (A) y el protón parcialmente expuesto está enlazado al dador (o dador) del enlace de hidrógeno (H-D).
¿Por qué hidrógeno? El hidrógeno es especial porque es el único átomo que (i) forma enlaces covalentes δ con átomos electronegativos como N, O y S, y (ii) usa el electrón (o los electrones) de la capa interior (1S) para el enlace covalente. Cuando su compañero de enlace electronegativo aleja a los electrones del hidrógeno, el núcleo del hidrógeno (un protón) queda expuesto en la parte lateral (distante del compañero enlazante). La parte desapantallada del protón queda expuesta, atrayendo la carga parcial negativa de un par solitario de electrones. El hidrógeno es el único átomo que expone su núcleo de ese modo. Otros átomos tienen electrones no-enlazantes localizados que apantallan el núcleo.
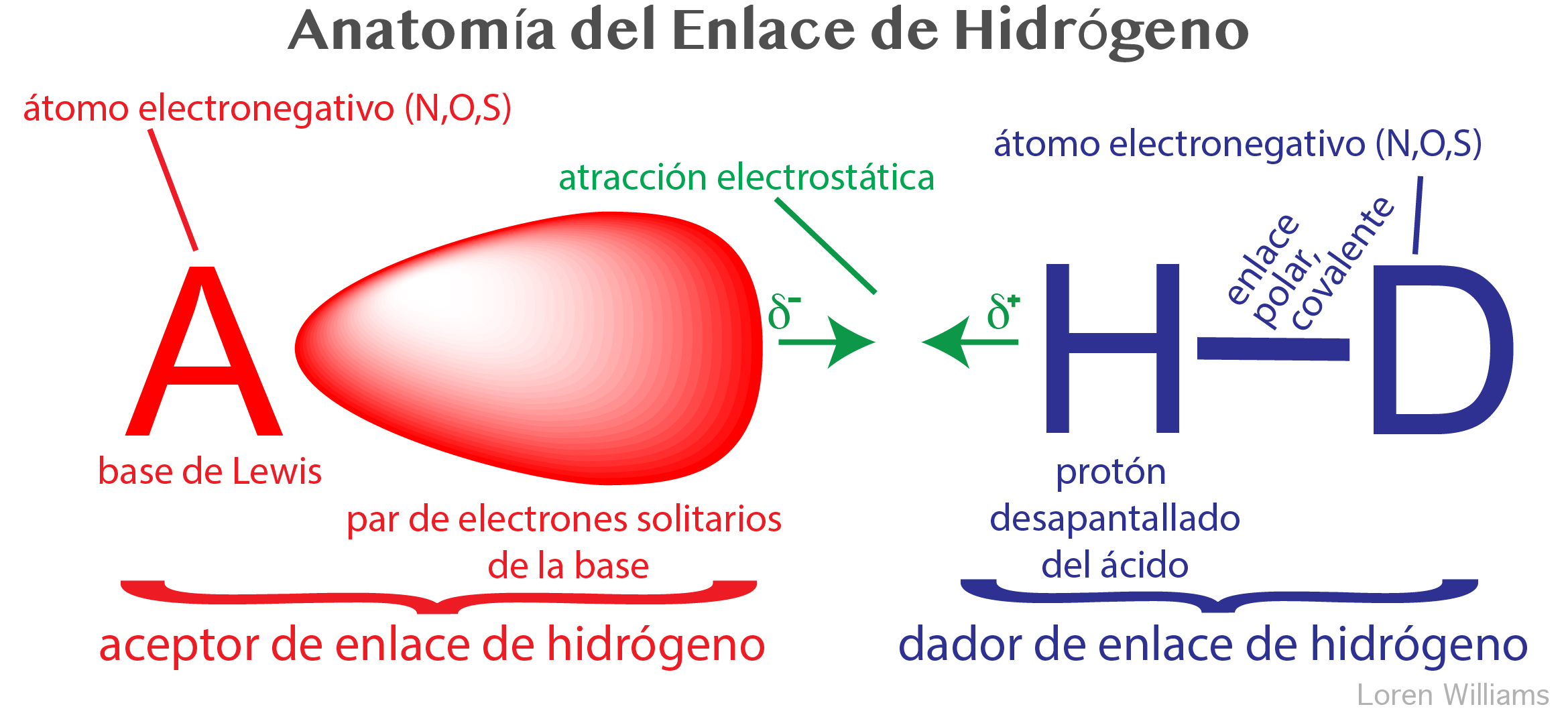
La figura 15 ilustra los elementos de un enlace de hidrógeno, incluyendo el aceptor HB y el dador HB, el par solitario y el protón expuesto. N, O, S son los átomos de enlace de hidrógeno predominantes (A y D) en los sistemas biológicos.
Un enlace de hidrógeno no es una reacción acido-base, donde el protón (H+) es completamente transferido de H-D a A para formar D- y HA+. Sin embargo, la fuerza de un enlace de hidrógeno correlaciona bien con la acidez del dador H-D y la basicidad del aceptor A. En un enlace de hidrógeno, el H+ es parcialmente transferido de H-D a A, pero el H+ se mantiene covalentemente enlazado a D. El enlace H-D permanece intacto.
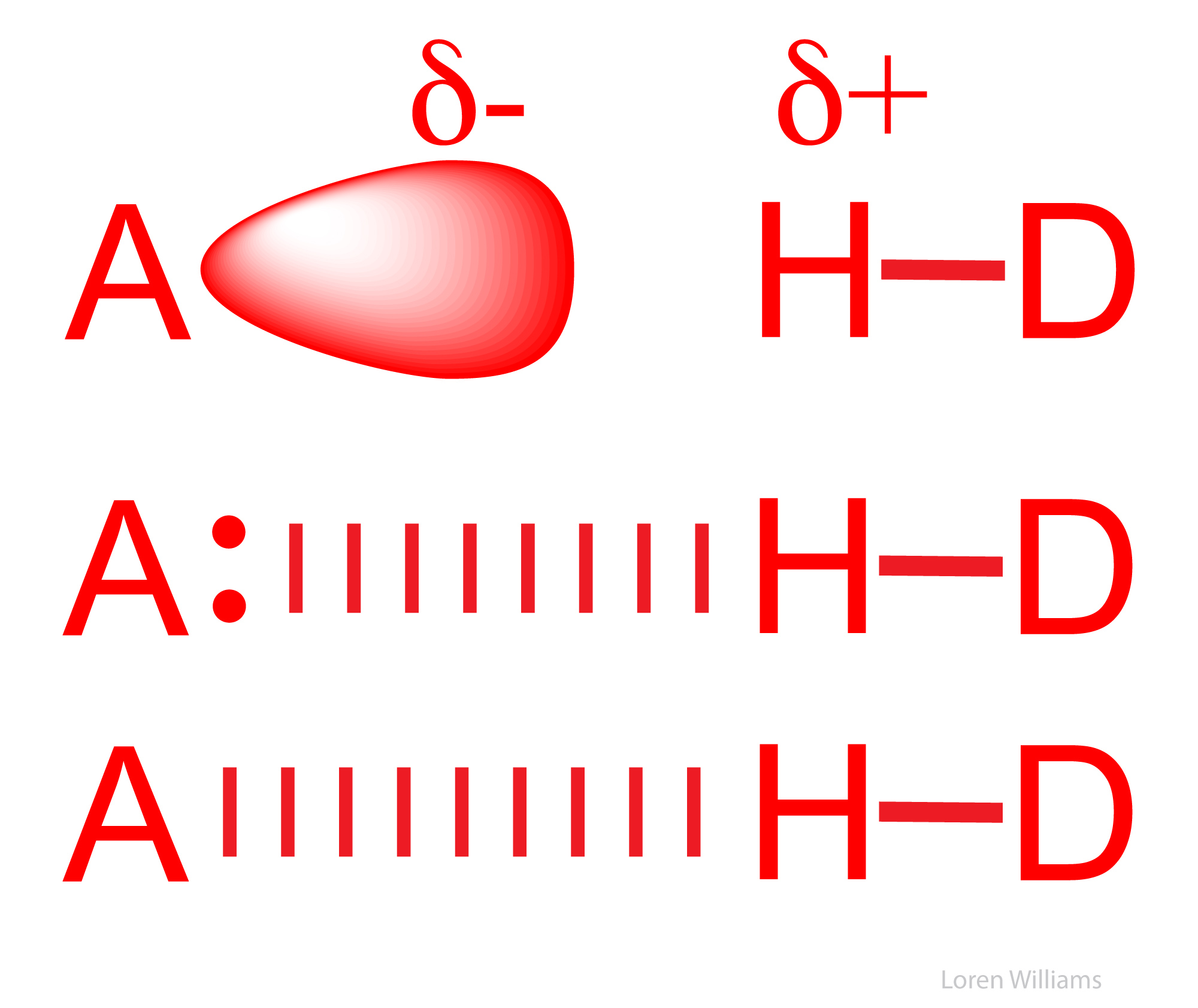
La figura 16 ilustra tres formas diferentes de representar un enlace de hidrógeno. El átomo A es la base de Lewis (por ejemplo, el N en NH3 o el O en H2O) y el átomo D es electronegativo (por ejemplo, O, N o S). La nomenclatura convencional es confusa: un enlace de hidrógeno no es un enlace covalente.
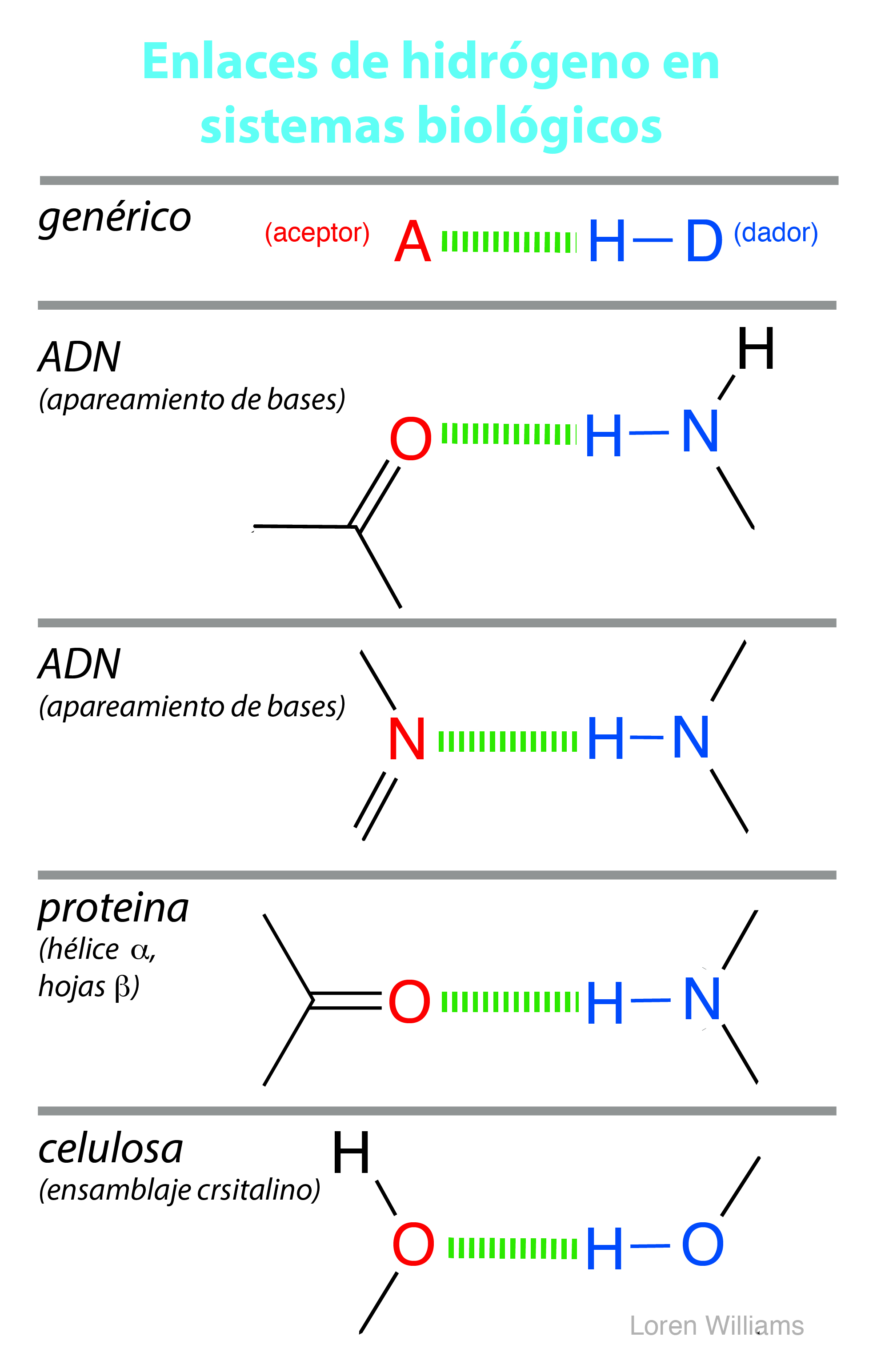
La figura 17 muestra los aceptores y dador de enlaces de hidrógeno más comunes en macromoléculas biológicas.
Los enlaces de hidrógeno más comunes en sistemas biológicos involucran átomos de oxígeno y de nitrógeno como A y D. Los grupos ceto (=O), amina (R3N), imina (R=N-R) y grupos hidroxilo (-OH) son los aceptores de enlace de hidrógeno más comunes en ADN, ARN, proteínas y carbohidratos más complejos. Los grupos hidroxilo y las aminas/iminas son los dadores de enlace de hidrógeno más comunes. Los grupos hidroxilo y las aminas/iminas pueden tanto donar como aceptar enlaces de hidrógeno.
Al recorrer la tabla periódica, el aumento de electronegatividad del átomo D desprovee de densidad electrónica al protón (en H-D), aumentando su carga parcial positiva, y aumentando la fuerza de cualquier enlace de hidrógeno. Los tioles (-SH) pueden donar y aceptar enlaces de hidrógeno, pero éstos son generalmente débiles, porque el sulfuro no es suficientemente electronegativo. Los enlaces de hidrógeno que involucran carbono, donde H-D corresponde a H-C, son observados, aunque son débiles e infrecuentes. El carbono no es lo suficientemente electronegativo para formar buenos enlaces de hidrógeno. Los enlaces de hidrógeno son esencialmente electrostáticos por naturaleza, aunque la energía puede ser descompuesta en contribuciones adicionales de polarización, intercambio repulsivo, transferencia de carga y mezcla.
La fuerza de los enlaces de hidrógeno forma un continuo. Los enlaces de hidrógeno fuertes de 20-40 kcal/mol (82-164 kJ/mol), formados generalmente entre dadores y aceptores cargados, son casi tan fuertes como los enlaces covalentes. Los enlaces de hidrógeno débiles de 1-5 kcal/mol (4-21 kJ/mol), a veces formados con el carbono como dador de protón, no son más fuertes que las interacciones dipolo-dipolo convencionales. Los enlaces de hidrógeno moderados, que son los más comunes, se forman entre dadores y aceptores neutros y son de 3-12 kcal/mol (12-50 kJ/mol).
Nota sobre la nomenclatura: Un enlace de hidrógeno no es un enlace. Es una interacción molecular (una interacción no enlazante). El desafortunado nombre que se le dio a esta interacción molecular hace tiempo, ha causado y continuará causando todo tipo de confusión. No hay que confundir los enlaces de hidrógeno con enlaces reales. No son lo mismo.
La geometría de un enlace de hidrógeno puede ser descrita por tres valores: la distancia de D a H, la distancia de H a A y el ángulo entre D, H y A. Las distancias dependen de los tipos de átomos A y D. Si ambos A y D son átomos de oxígeno, entonces óptimamente, de H a A = 1.8 Å y D a H = 1.0 Å. Los enlaces de hidrógeno más estables están cerca de ser lineares (ángulo entre D, H y A de 180°). Los enlaces de hidrógeno en hojas β antiparalelas son lineares, mientras que los enlaces de hidrógeno en hojas β paralelas son no-lineares.
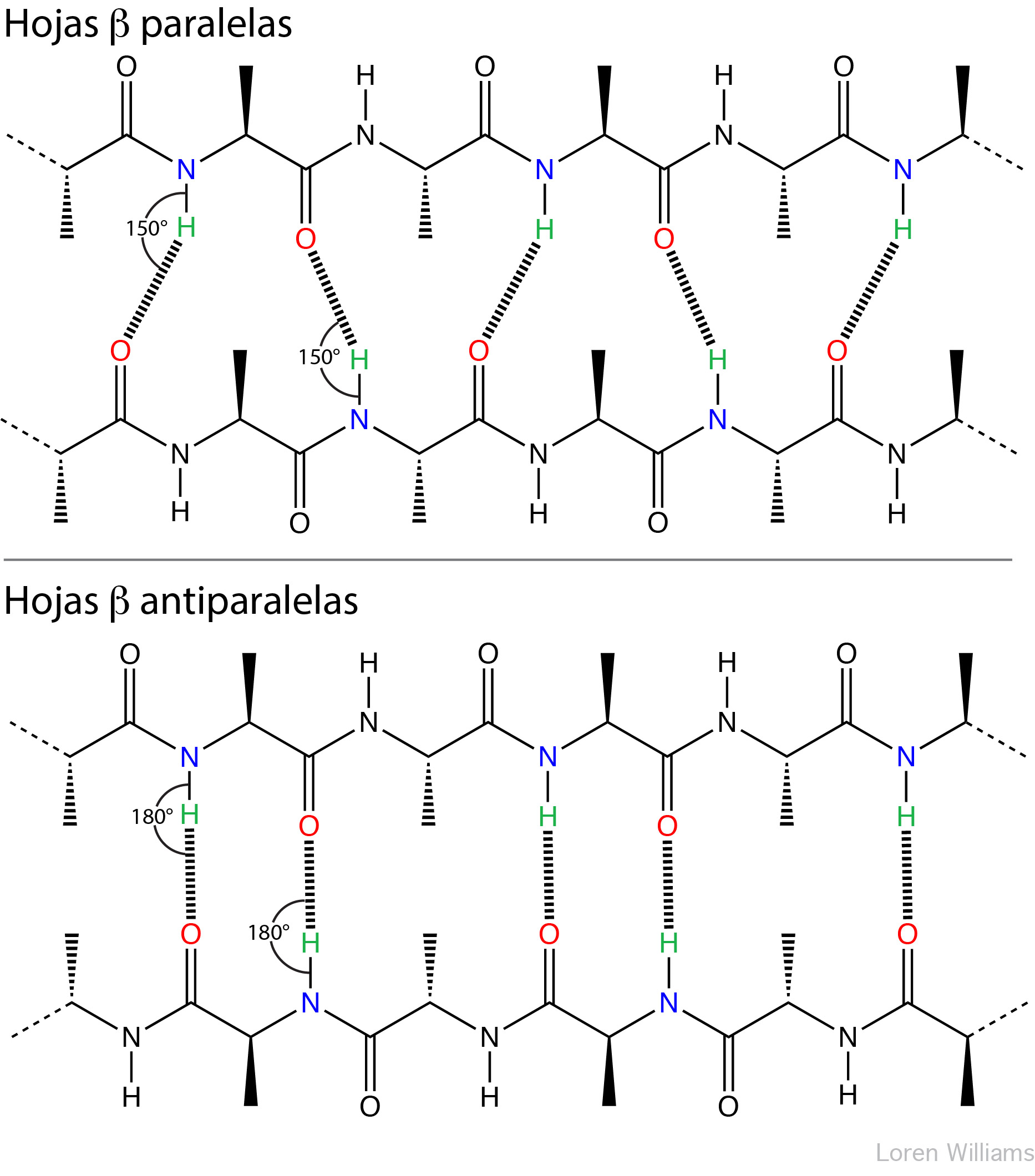
La Figura 18 ilustra la no linealidad de los enlaces de hidrógeno de la hoja β paralela y la linealidad de los enlaces de hidrógeno de la hoja β antiparalela.
Los enlaces de hidrógeno pueden tener dos centros (como en las hojas β y la estructura cristalina hexagonal del hielo), tres centros o cuatro centros. Los enlaces de hidrógeno de dos centros son generalmente cortos, más lineares, y fuertes que los enlaces de hidrógeno de tres y cuatro centros. Los enlaces de hidrógeno de tres centros a veces se llaman bifurcados mientras que los enlaces de hidrógeno con cuatro centros se llaman a veces trifurcados.
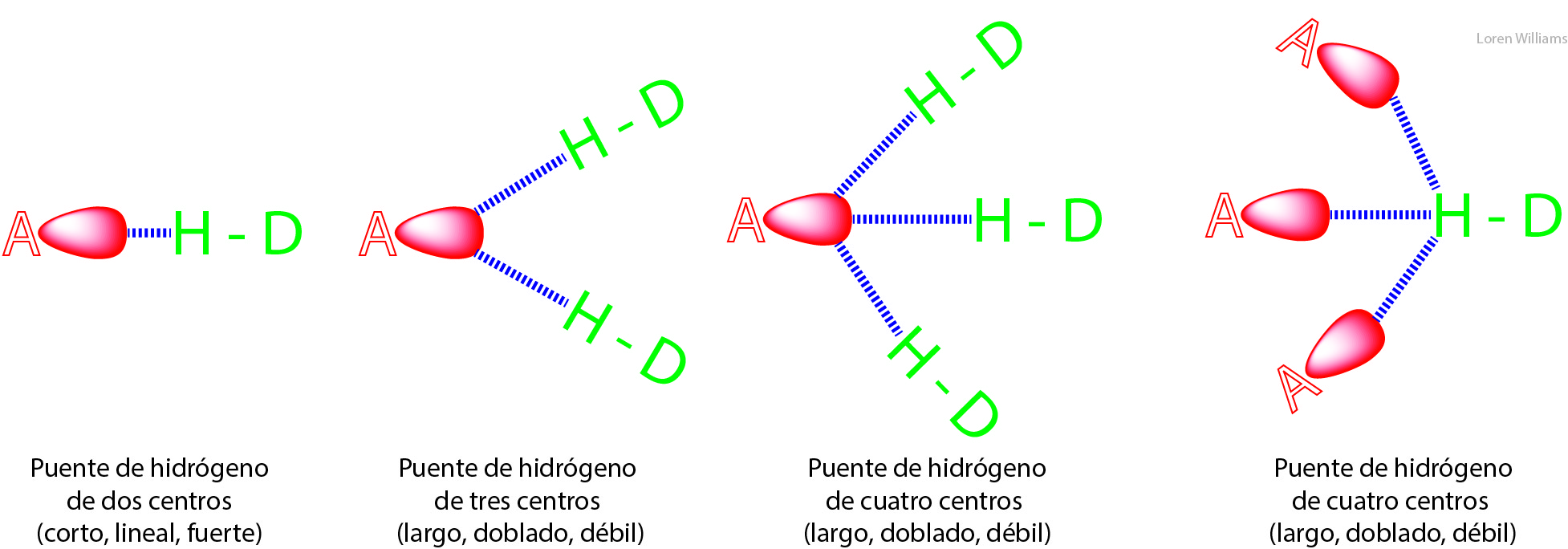
La figura 19 ilustra enlaces de hidrógeno de dos, tres y cuatro centros. El enlace de hidrógeno de dos centros es el más cercano a un enlace de hidrógeno "ideal" y es más fuerte que los otros tipos. El esquema de enlace de hidrógeno de cuatro centros a la derecha se puede observar en el amonio cristalino, donde un solo par aceptor tiene que acomodar a tres dadores (ver la sección sobre amoníaco, a continuación).
Los átomos de hidrógeno no son observables mediante cristalografía de rayos x como ocurre con las proteínas y los ácidos nucleicos. De modo que una descripción geométrica del enlace de hidrógeno que sea dependiente de la posición del protón no es práctica en estructuras proteicas o de ácidos nucleicos. En esos casos, uno está generalmente limitado al análisis de la distancia de dador a aceptor. Es común atribuir la presencia de un enlace de hidrógeno si la distancia entre aceptor y dador es menor que la suma de sus radios de van der Waals. Aunque probablemente este límite es muy conservador. El mejor criterio para un enlace de hidrógeno es que haya una distancia menor de 3.4 Å entre D y A.
En sistemas biológicos, los enlaces de hidrógeno son frecuentemente cooperativos y se estabilizan por resonancia involucrando varios enlaces de hidrógeno. En sistemas con múltiples enlaces de hidrógeno, la fuerza de un enlace de hidrógeno es aumentada por un enlace de hidrógeno adyacente. Por ejemplo, en los sistemas de enlace de hidrógeno de abajo (el dímero de ácido acético), el enlace de hidrógeno de arriba aumenta tanto la acidez del hidrógeno, como la basicidad del oxígeno en el enlace he hidrógeno de abajo. Cada enlace de hidrógeno hace al otro más fuerte de lo que sería en aislamiento. La cooperatividad del enlace de hidrógeno se observa en el emparejamiento de bases de ácidos nucleicos y en el plegamiento de proteínas.
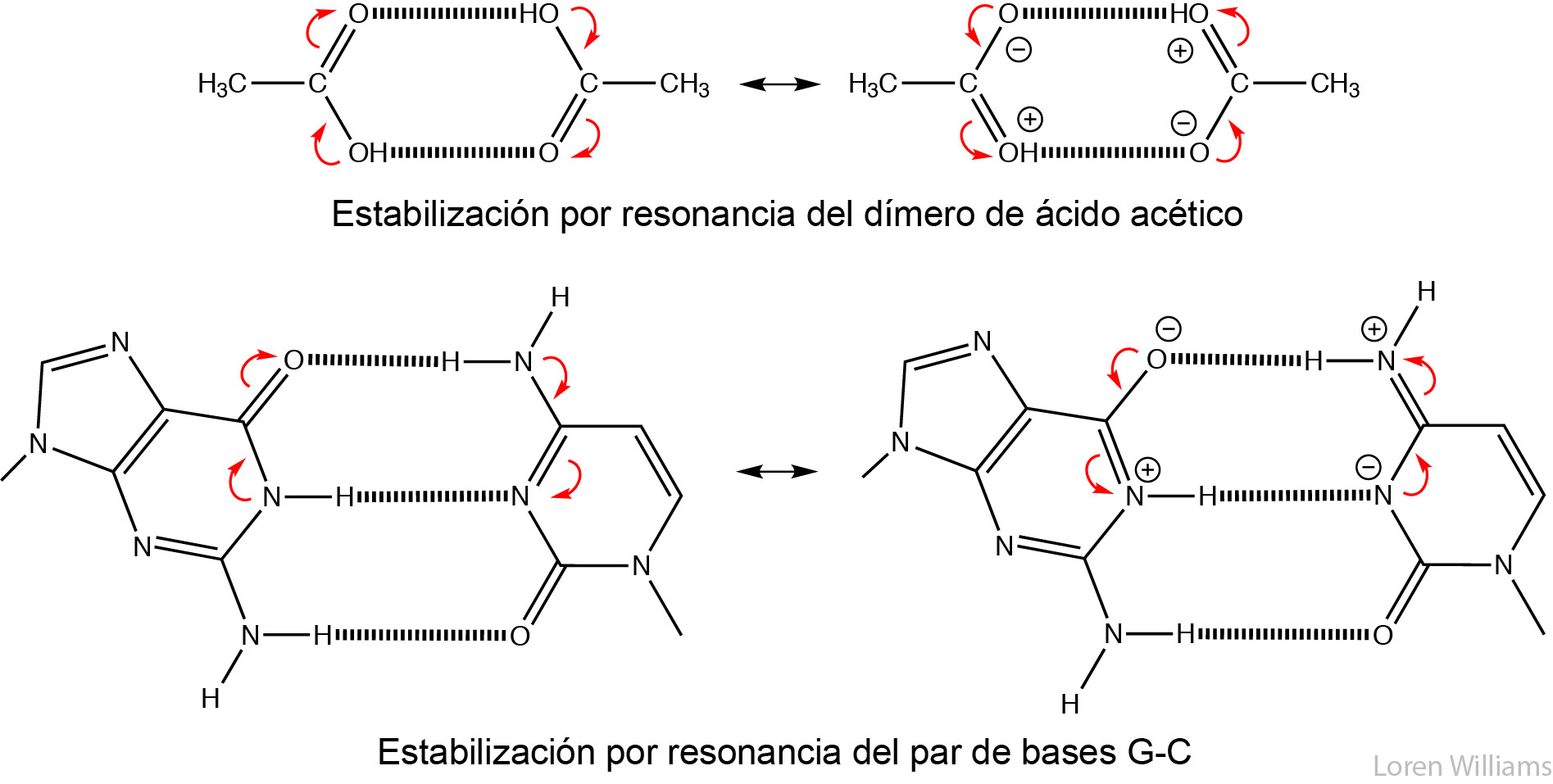
La figura 20 muestra la cooperatividad de los enlaces de hidrógeno de un dímero de ácido acético (arriba) y de un par de bases G-C (abajo). Un enlace de hidrógeno aumenta la estabilidad del enlace de hidrógeno adyacente (y viceversa).
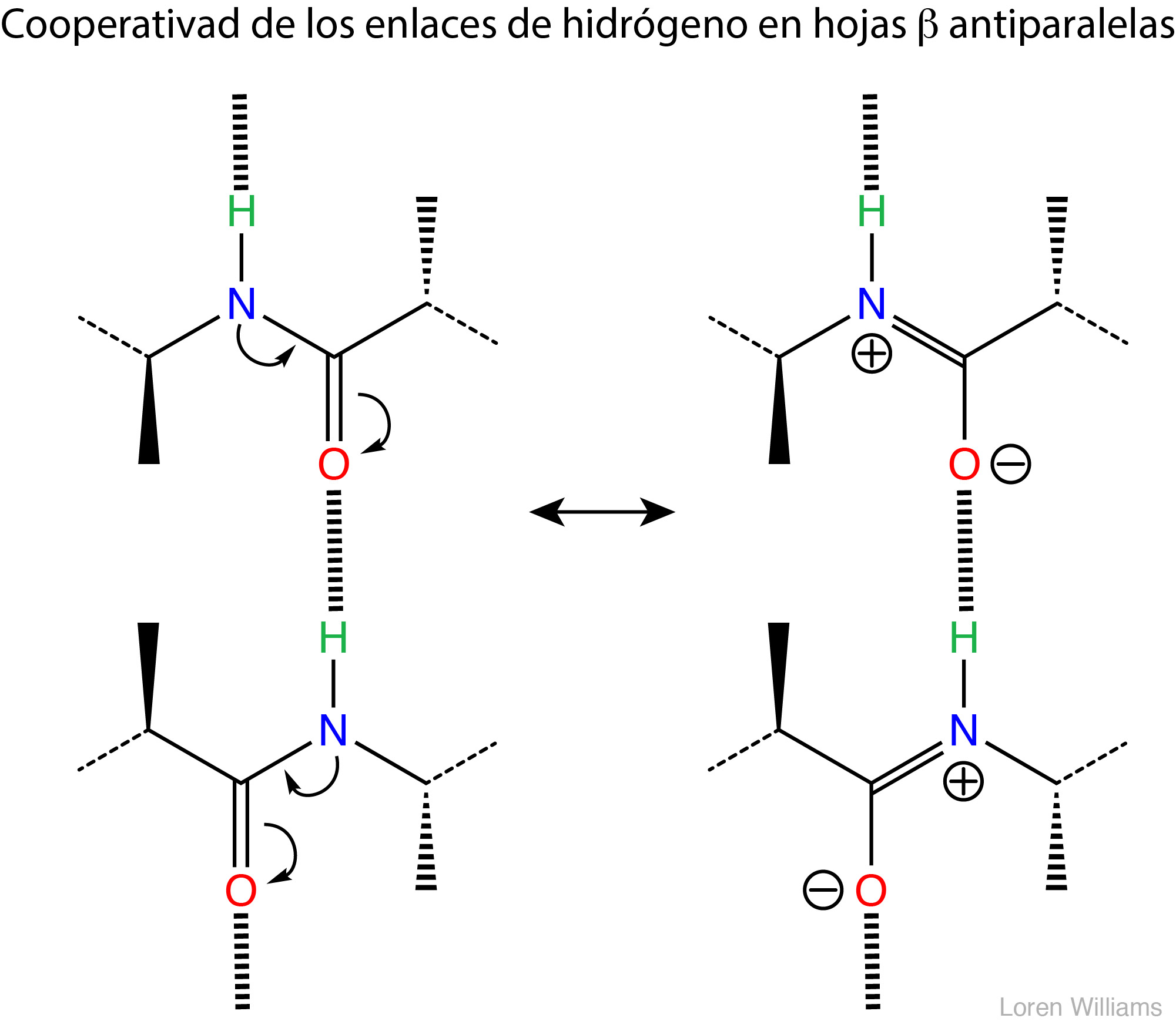
La figura 21 muestra cooperatividad a través de resonancia de los enlaces de hidrógeno de una hoja β anti paralela.
La vida tal y como la conocemos en la Tierra depende completamente del agua. El agua es el medio de la biología; alrededor del 80% del peso de los organismos vivos es agua.
Aunque no es generalmente descrita como tal, el agua es el metabolito universal más frecuente en química biológica. Un metabolito es el reactivo/intermedio/producto de una reacción molecular de bajo peso molecular. Un humano utiliza una cantidad de moléculas de agua superior a su masa corporal cada día.
El uso de agua como metabolito se da en la formación de biopolímeros. Todos los biopolímeros se forman por reacciones de condensación y deshidratación, que unen los pequeños componentes básicos y producen agua químicamente. Específicamente, un enlace peptídico en una proteína se forma por condensación de amino ácidos. En la reacción neta, dos amino ácidos se unen y producen una molécula de agua para formar en enlace peptídico. El agua es un producto de la reacción química de formación de enlace peptídico. En la reacción inversa, los biopolímeros se degradan por reacciones de hidrolisis, que químicamente consumen agua. El agua es el reactivo en la reacción de ruptura de un enlace peptídico.
Los polinucleótidos (ADN y ARN) se forman por condensación de nucleótidos (dG, dT, dA, dC en ADN), que a su vez son formados por condensación de subestructuras mas pequeñas. Los triglicéridos y fosfolípidos se forman por condensación de glicerol con ácidos grasos y otras moléculas. La celulosa, el polímero mas abundante en la biosfera, se forma por condensación de glucosa.
En resumen: el agua es el medio de la biología (el solvente) y esta totalmente integrado en las reacciones químicas universales mas básicas de la biología. Mantente hidratado.
El agua es intrinsecamente auto-complementaria. En agua líquida o sólida, los tres átomos de cada molécula de agua participan en enlaces de hidrógeno casi ideales. Los dadores y aceptores de cualquier molécula de agua encajan geométricamente con los dadores y aceptores de moléculas de agua cercanas. El agua líquida y solida tiene mayor densidad de enlaces de hidrógeno ideales (por volumen) que cualquier otro material. En la fase condensada del agua (líquida o sólida), los grupos enlazantes de hidrogeno de cada molécula de agua son complementarios a los grupos enlazantes de hidrógeno del agua circundante.
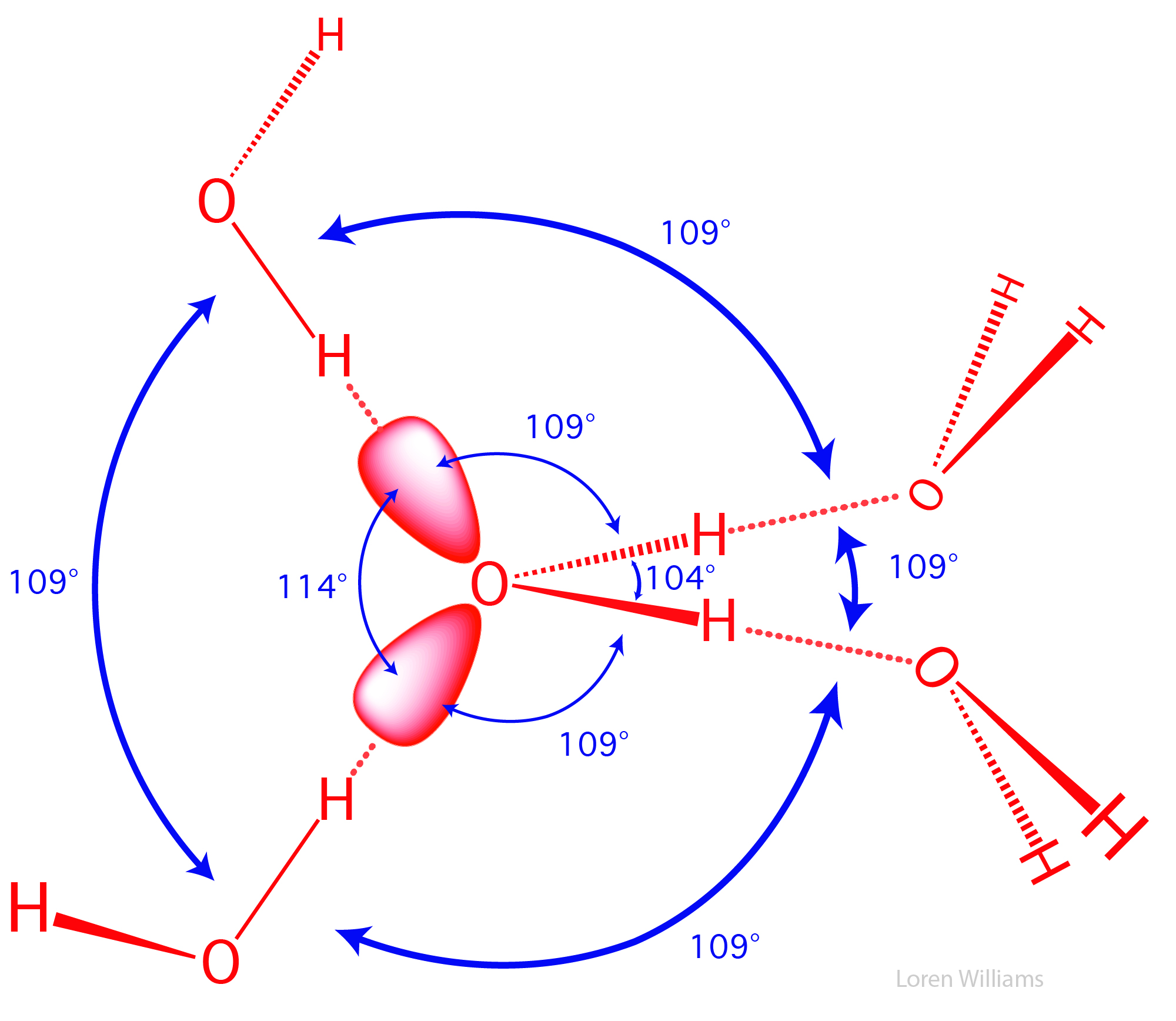
La figura 21a ilustra la complementariedad de las interacciones de enlace de hidrógeno de una molécula de agua con el entorno en agua líquida o sólida. El anillo interior de ángulos está dentro de una molécula de agua. El anillo exterior de ángulos se encuentra entre las moléculas de agua circundantes.
El agua tiene el mismo número de enlaces de hidrógeno aceptores y dadores (dos de cada). En fases condensadas, cada molécula de agua actúa como dador en dos enlaces de hidrógeno y como aceptor en otros dos. La auto-complementariedad del agua es emergente en la fase condensada. Moléculas de agua aisladas o en pequeños grupos no muestran auto-complementariedad.
La fuerte cohesión entre moléculas de agua causa altos puntos de fusión, ebullición, calor de vaporización, calor de fusión y tensión superficial. El agua crece (aumenta su volumen) al congelarse; el hielo flota. El calor de vaporización del agua (540 cal/g) es unas dos veces mayor que el del etanol (263 cal/g) y casi diez veces mayor que el del cloroformo (59 cal/g).
El agua es un buen solvente para iones y sustancias polares y un mal solvente para sustancias no polares. El agua hace que ciertas moléculas anfipáticas (con funcionalidades tanto polares como no polares) formen compartimentos espontáneamente. En el agua, las membranas se forman y las proteínas se pliegan.
El agua tiene la singular característica de apantallar la interacción entre especies cargadas. Las interacciones electrostáticas entre iones están fuertemente atenuadas en el agua. La fuerza electrostática entre dos iones en solución es inversamente proporcional a la constante dieléctrica del solvente. La constante dieléctrica del agua (80.0) es enorme. Es más del doble que la del metanol (33.1) y más de cinco veces la del amonio (15.5). El agua es un buen solvente para las sales porque las fuerzas de atracción entre cationes y aniones son minimizadas por el agua.
Una molécula de agua (H2O) aislada puede formar fuertes enlaces de hidrógeno tanto con dadores de enlace de hidrógeno como con aceptores.
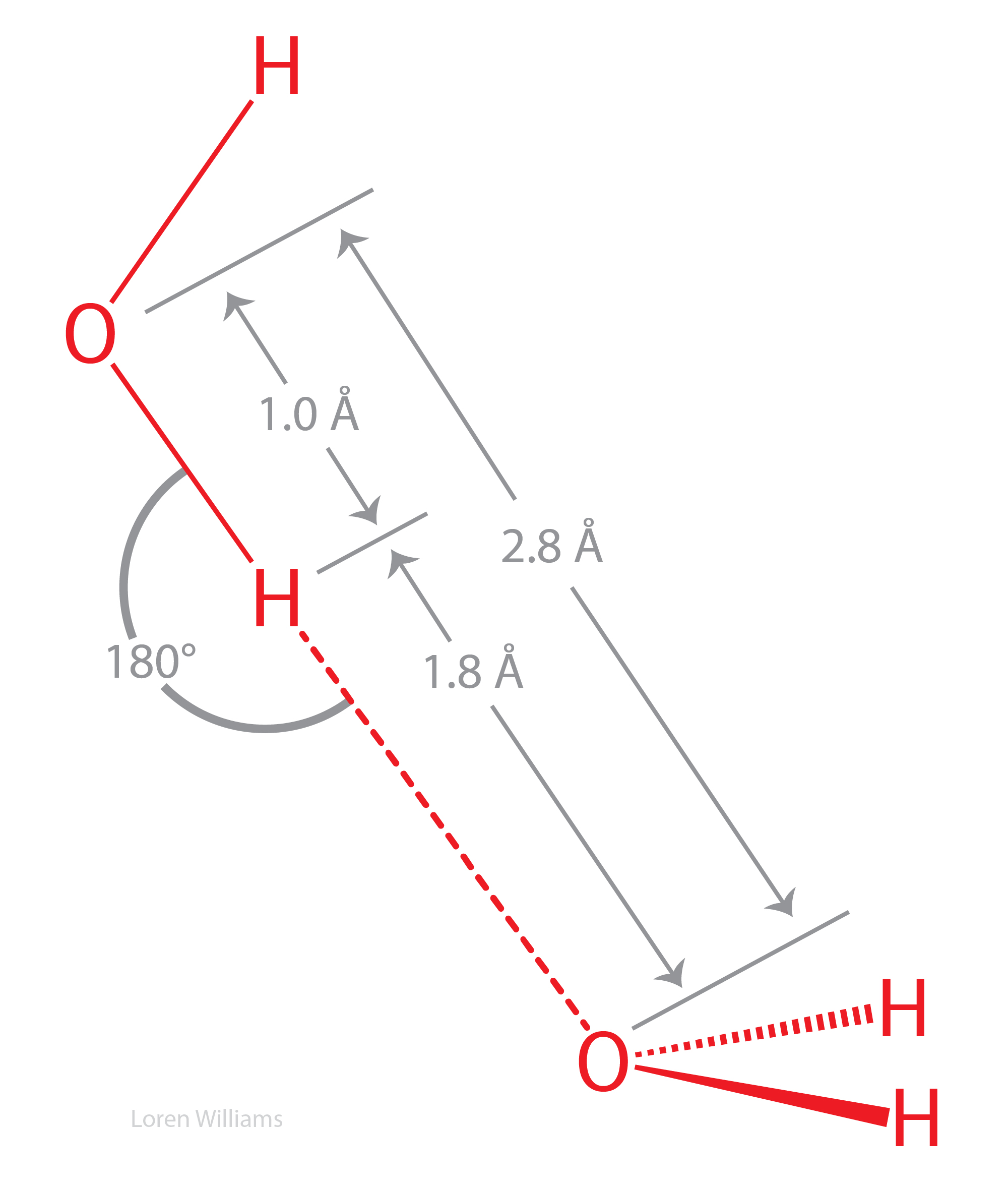
La Figura 22 ilustra el enlace de hidrógeno entre dos moléculas de agua. Los enlaces de hidrógeno son cortos, lineales y fuertes. Estos son enlaces de hidrógeno de dos centros. Aunque cada molécula de agua en el agua líquida y en el hielo forma cuatro enlaces de hidrógeno, aquí solo se muestra un enlace de hidrógeno.
Los enlaces de hidrógeno provocan la violación del principio de las superficies de van der Waals. La distancia del enlace de hidrógeno entre H y O está en torno a 1.8 Å, que es menor que la suma de los radios de van der Waals para O y H (rO=1.5 Å; rH=1.0 Å). Nótese que la distancia de enlace de hidrógeno entre O y O está en torno a 2.8 Å, que es menor que dos veces el radio de van der Waals del oxígeno (rO=1.5 Å).
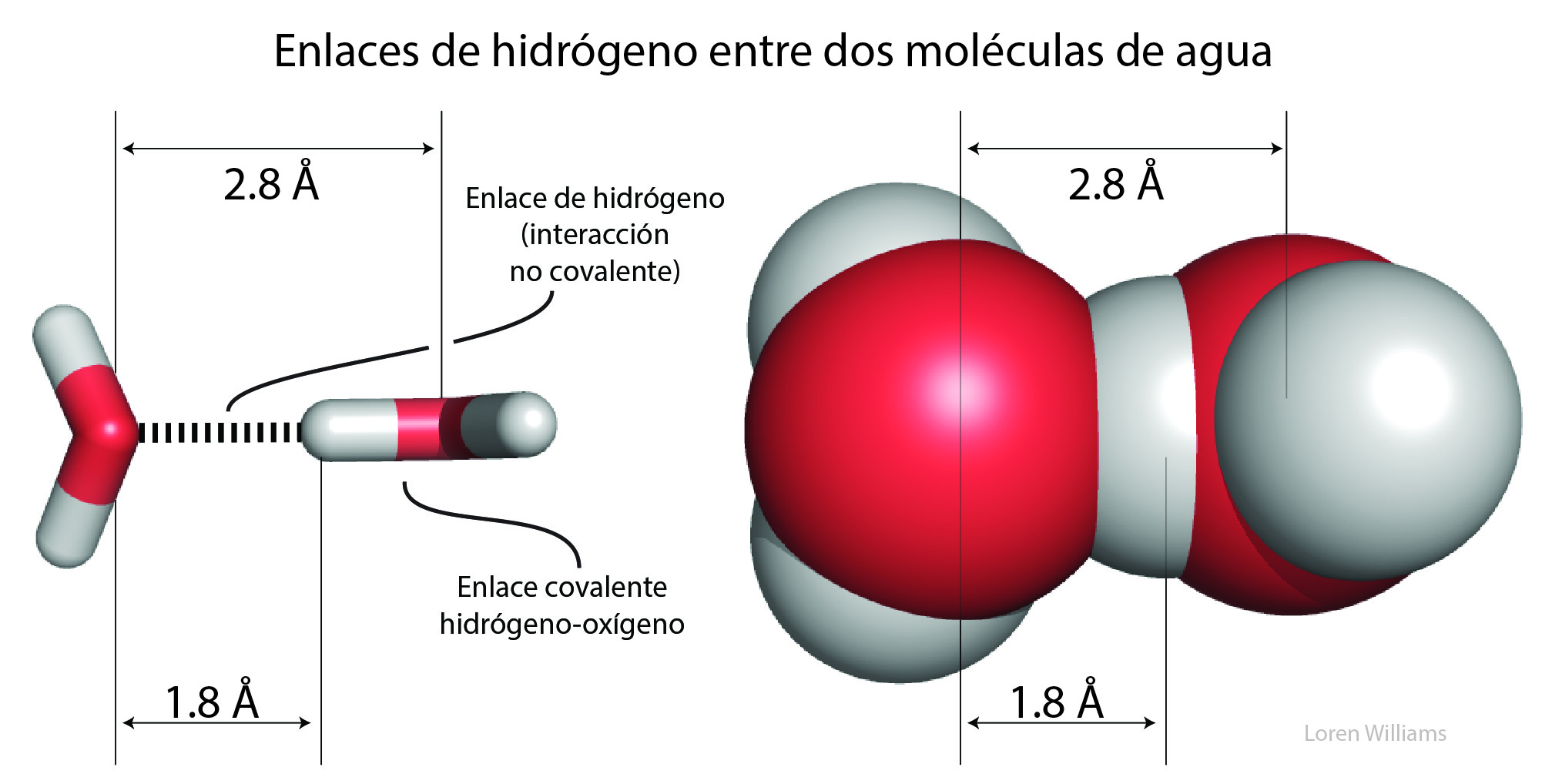
La figura 23 muestra cómo los enlaces de hidrógeno unen dos moléculas de agua. Esta figura ilustra la diferencia entre un enlace covalente, que une un átomo de oxígeno a un átomo de hidrógeno, y un enlace de hidrógeno, que también une un oxígeno a un hidrógeno. Un enlace de hidrógeno es una interacción molecular no covalente. Los átomos de oxígeno están representados en rojo y los de hidrógeno en blanco. La representación de la derecha muestra cómo los enlaces de hidrógeno provocan la violación de las superficies de van der Waals.
El oxígeno es altamente electronegativo, y gana carga parcial negativa a través de la captación de densidad de carga electrónica de los dos átomos de hidrógeno a los cuáles está enlazado covalentemente, dejándolos con cargas parciales positivas. El agua es un excelente solvente de enlaces de hidrógeno. Las coordenadas de una molécula de agua unida por enlaces de hidrógeno a otras dos moléculas de agua están aquí [coordenadas].
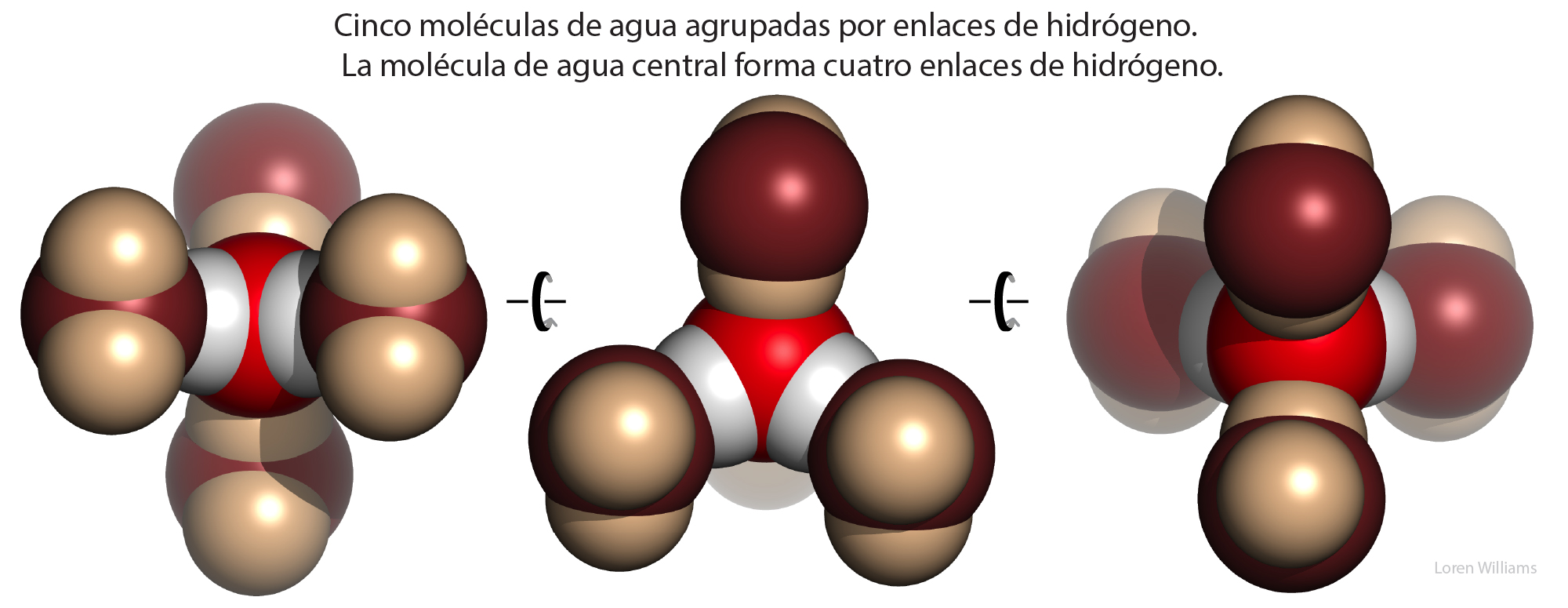
La figura 24 ilustra como una molécula de agua puede donar dos enlaces de hidrógeno y aceptar dos enlaces de hidrógeno. La molécula de agua central está donando dos enlaces de hidrógeno y aceptando dos enlaces de hidrógeno. En agua líquida, el número total de dadores de enlaces de hidrógeno es igual al número total de aceptores de enlaces de hidrógeno. Todos los dadores y aceptores de enlaces de hidrógeno están satisfechos. El agua es auto-complementaria.
Las coordenadas de un cubo de hielo muy pequeño están aquí [coordenadas]. Para información adicional sobre el agua, ver la sección H6. Descripción empírica del efecto hidrofóbico.
El oxígeno de una molécula de agua tiene cuatro orbitales de valencia llenos (sp3 hibridados) que forman un tetraedro moderadamente distorsionado. Dos de los pares de electrones forman enlaces covalentes con átomos de hidrógeno y los otros dos no están enlazados. Los pares solitarios (o desapareados) no enlazados ocupan más espacio que los pares enlazados, causando la distorsión de un tetraedro perfecto. Es útil imaginar que una molécula de agua es un tetraedro con carga negativa en dos vértices y carga positiva en otros dos.
El oxígeno, que es altamente electronegativo, atrae la densidad electrónica de los átomos de hidrógeno hasta el punto de que se convierten prácticamente en protones en sus lados expuestos (opuestos al oxígeno). La distribución de carga de una molécula de agua (carga parcial negativa en el oxígeno y carga parcial positiva en el hidrógeno) se muestra a continuación:
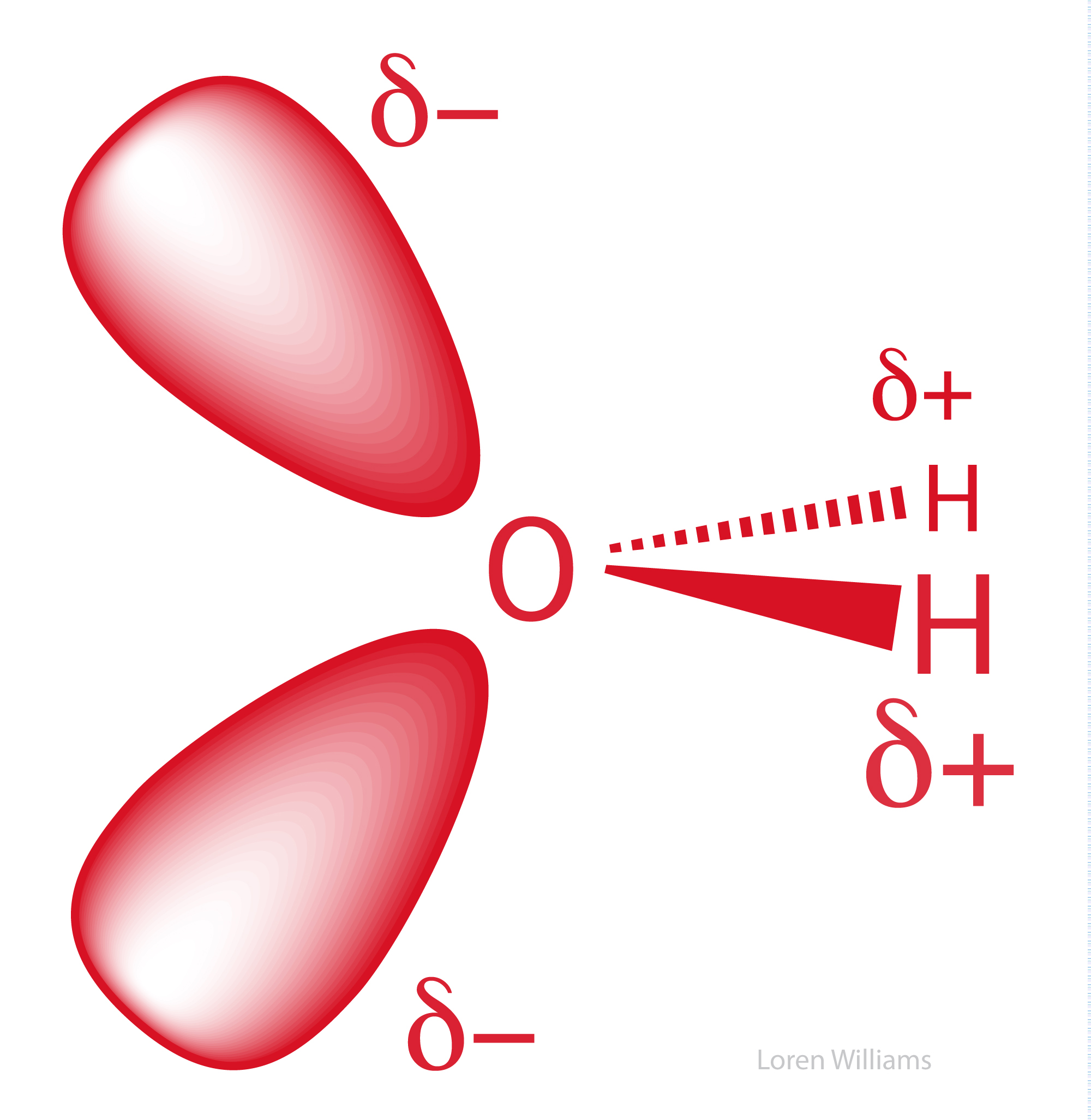
La figura 25 ilustra los dos pares de electrones solitarios y los dos pares de electrones enlazados de una molécula de agua. Cuatro orbitales de valencia de una molécula de agua forman un tetraedro ligeramente distorsionado. Los pares de electrones no enlazados ocupan un poco más de espacio que los pares de electrones enlazados.
Mediante rayos X y difracción de neutrones se observa que en el hielo cristalino cada molécula de agua forma cuatro enlaces de hidrógeno con distancias intermoleculares oxígeno-oxígeno de 2.76 Å. Cada átomo de oxígeno está localizado en el centro de un tetraedro formado por otros cuatro átomos de oxígeno. Cada átomo de hidrógeno se encuentra alineado entre dos átomos de oxígeno y forma un enlace covalente con uno de ellos (longitud del enlace: 1.00 Å) y un enlace de hidrógeno con el otro (longitud del enlace de hidrógeno: 1.76 Å). La forma tetraédrica de una molécula individual de agua es proyectada hacia la red cristalina circundante. Los átomos de hidrógeno no se encuentran en el punto medio entre átomos de oxígeno. Para más información consulte la sección G. Enlaces de hidrógeno..
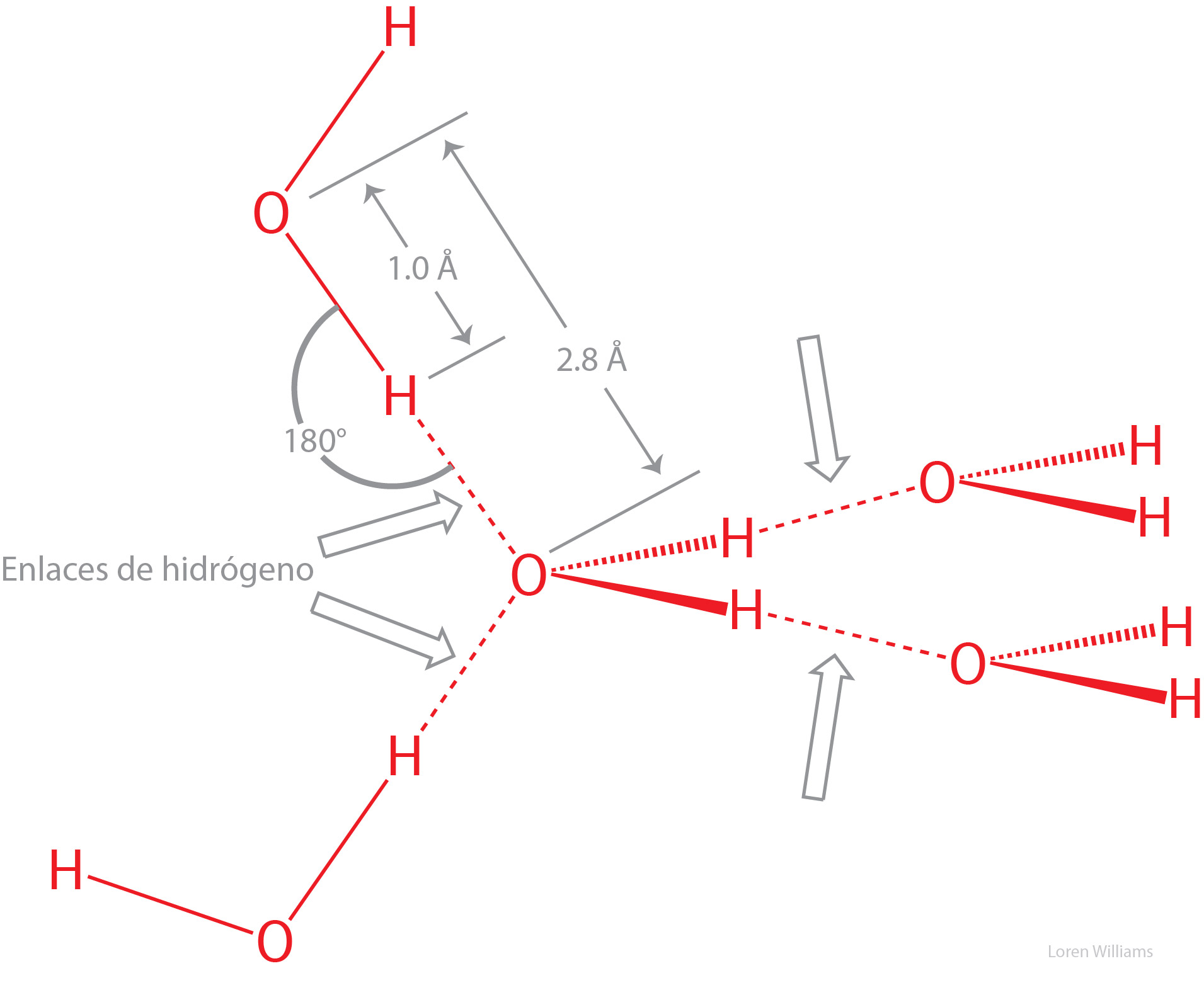
La figura 26 muestra las interacciones de enlaces de hidrógeno de una molécula de agua con otras cuatro moléculas en agua líquida o sólida. Los grupos dadores y aceptores de una molécula de agua dada son complementarios a los grupos dadores y aceptores colectivos de las moléculas de agua circundantes. Una molécula de agua puede donar dos enlaces de hidrógeno y aceptar otros dos.
Las moléculas de agua en estado cristalino no están empaquetadas muy densamente, lo que resulta en pequeñas cavidades de espacio vacío dentro del cristal. Las pequeñas cavidades en la red cristalina, pero no en el líquido, son la razón por la cual el agua aumenta de volumen al congelarse (y por la que el hielo flota). Hay muchos grados de libertad en las relaciones dador/aceptor de enlaces de hidrógeno que son inter-convertidos mediante rotaciones cooperativas. Las moléculas de agua giran fácilmente en hielo.
Se puede aprender mucho sobre el agua (H2O) estudiando el amoníaco (NH3). Una comparación de amoníaco con agua muestra la importancia de la auto-complementariedad del agua, donde las geometrías de los dadores y aceptores de enlace de hidrógeno de cualquier molécula de agua dada complementan las de las moléculas de agua circundantes. Una molécula de amoníaco no es complementaria, con tres sitios dadores (N-H) y un sitio aceptor.
Una molécula de amoníaco aislada, al igual que una molécula de agua, puede formar fuertes enlaces de hidrógeno con dadores o aceptores de enlaces de hidrógeno. El amoníaco es más básico que el agua y, por lo tanto, el amoníaco es un mejor aceptor de enlaces de hidrógeno que el agua.
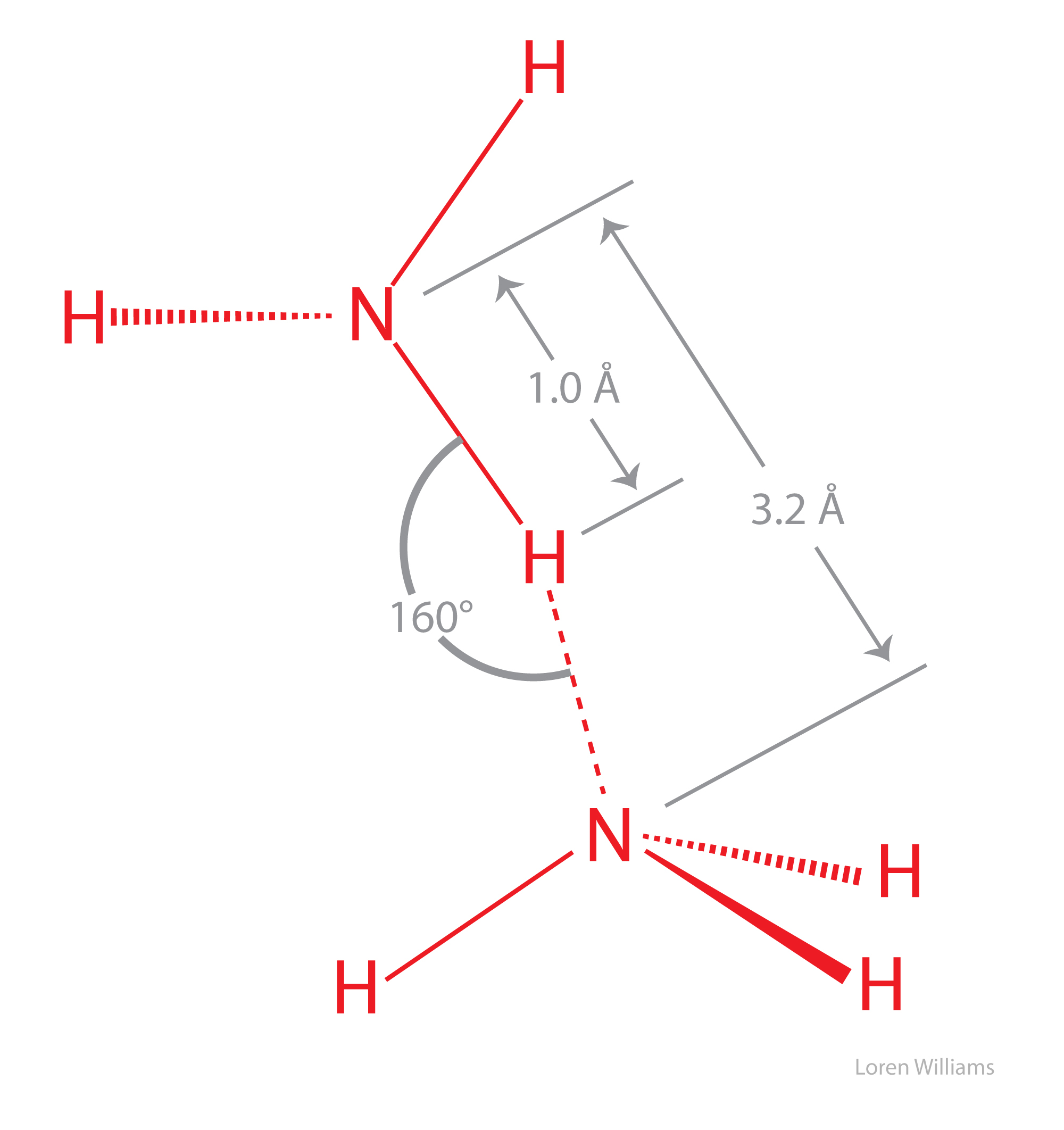
La figura 27 ilustra el enlace de hidrógeno como se observa en el amoníaco cristalino. Los enlaces de hidrógeno son más largos que los del hielo y no son lineares. Aunque cada molécula de amoníaco forma enlaces de hidrógeno con seis vecinos en el cristal, aquí solo se muestran dos moléculas de amoníaco.
En los estados cristalino y líquido, el único par de electrones en cada nitrógeno es compartido por múltiples dadores de enlaces de hidrógeno. Los enlaces de hidrógeno son bifurcados y trifurcados, como se describió anteriormente (ver figura 20). Los enlaces de hidrógeno en el estado cristalino y líquido son largos, no lineares y débiles.
El punto de ebullición del amoníaco es −33 °C, mucho más bajo que el del agua (100 °C), lo que indica que las interacciones moleculares en NH3 (liq) son significativamente más débiles que en H2O (liq). Puedes encontrar las coordenadas de una molécula de amoníaco aquí [coordenadas].
En el estado líquido, las moléculas de agua no se encuentran tan ordenadas como en el estado cristalino. En el estado líquido, a 0 °C, una molécula de agua participa en aproximadamente 3.5 enlaces de hidrógeno intermoleculares. Algunos de los enlaces de hidrógeno tienen tres o cuatro centros. El agua líquida es más densa que el agua sólida. A pesar de ello, las propiedades macroscópicas del agua líquida están dominadas por las interacciones cohesivas direccionales y complementares entre moléculas de agua.

El efecto hidrofóbico da lugar a la insolubilidad del aceite y otras sustancias no polares en agua. Si mezclas aceite y agua mediante agitación vigorosa, observarás una separación (inmiscibilidad) espontánea – lo que significa que la entropía de la mezcla es negativa. La mezcla espontánea es extraña e inusual. Si mezclas canicas rojas y azules, o agua y etanol, o N2 (g) y O2 (g), no observaras una inmiscibilidad espontánea, lo que significa que la entropía de la mezcla es positiva. La mezcla suele ser espontánea porque el número de estados accesibles aumenta con la mezcla. Se pueden dar más formas (o estados) en las sustancias en la mezcla que sin mezclar. La mezcla de sustancias no polares y agua es el efecto hidrofóbico en acción. Las sustancias hidrófobas son aquellas que son solubles en solventes no polares (como CCl4, ciclohexano o aceite de oliva). La definición excluye sustancias como la celulosa, que son insolubles debido a la fuerte cohesión intermolecular. Los hidrocarburos (CH3CH2CH2... CH2CH3) son hidrófobos.
El efecto hidrofóbico solo se puede entender después de pensar profundamente en el agua. Las fuerzas atractivas entre solutos no polares emanan del medio (agua), no de interacciones directas entre las moléculas de soluto. Las sustancias hidrofóbicas participan pasivamente en el fenómeno hidrofóbico. Las interacciones moleculares de un hidrocarburo con moléculas de agua en solución acuosa son tan favorables como con las moléculas de hidrocarburo vecinas en hidrocarburo líquido puro.
El efecto hidrofóbico es una consecuencia indirecta de las fuertes interacciones direccionales entre las moléculas de agua y la complementariedad de esas interacciones. El efecto hidrofóbico es una propiedad del agua; es consecuencia de la estructura molecular específica del agua y sus propiedades cohesivas únicas. (Muchos libros de texto contienen explicaciones superficiales o incorrectas que no explican la base del efecto hidrofóbico).
Podemos comprender las explicaciones moleculares y termodinámicas del efecto hidrofóbico de forma separada.
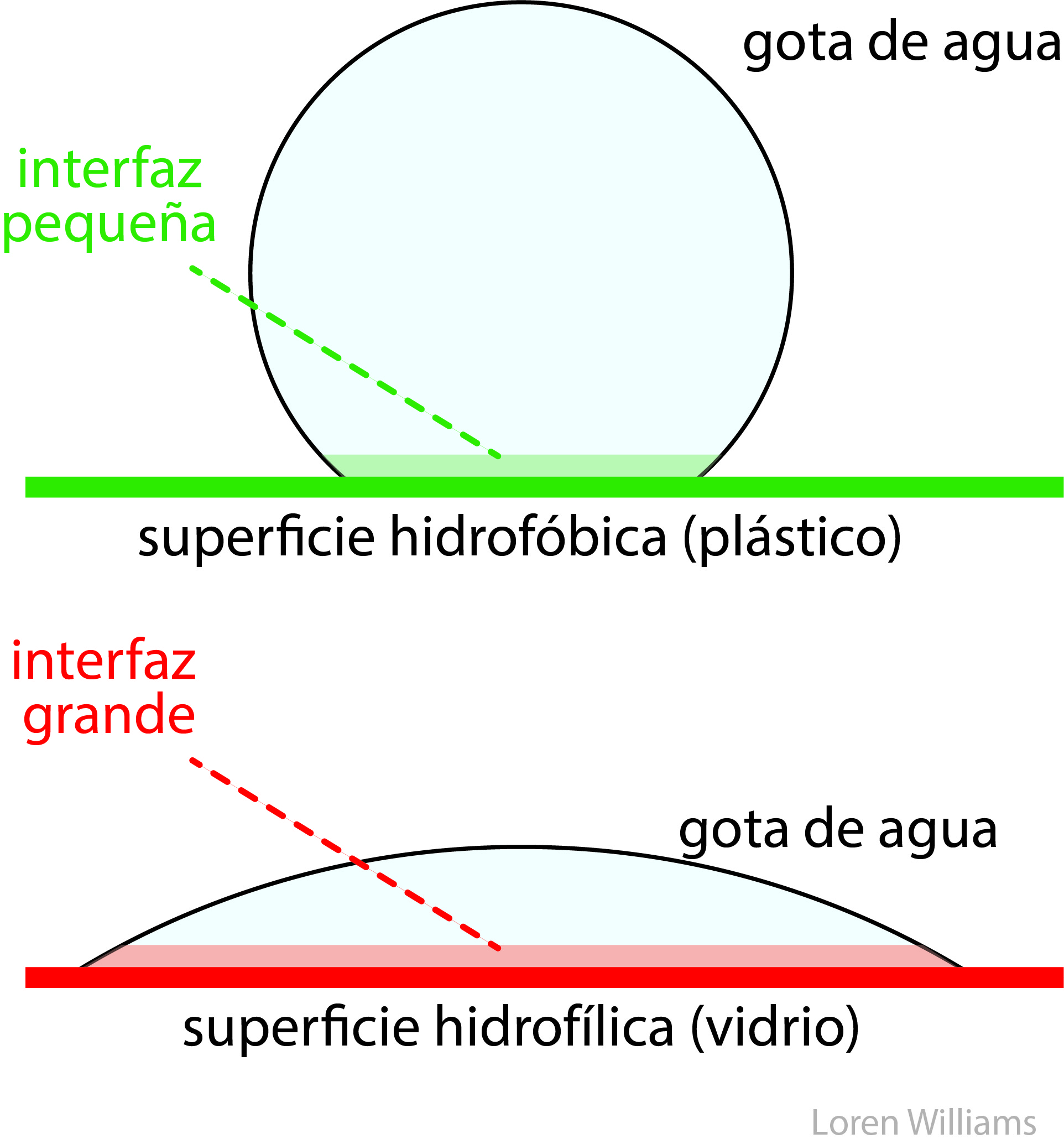
Las interacciones cohesivas entre las moléculas de agua no se interrumpen por la presencia de hidrocarburos disueltos (u otras moléculas no polares). Las interacciones entre las moléculas de agua en la región de interfaz (en contacto con los hidrocarburos) son tan extensas y favorables (en términos de entalpía) como las interacciones entre las moléculas de agua en el agua en conjunto, rodeadas solo de agua. No hay un cambio neto en la extensión de las interacciones moleculares favorables cuando el aceite y el agua se mezclan o se separan.
El impulsor de la separación son las moléculas de agua en la interfaz (directamente adyacentes a una molécula de hidrocarburo o superficie de plástico) que mantienen las interacciones agua-agua a costa de la libertad de rotación y traslación. El agua interfacial tiene baja entropía y, por lo tanto, su presencia es desfavorable. El agua gana entropía y, por lo tanto, estabilidad al minimizar la cantidad de agua interfacial. Esta es la razón por la cual las gotas de agua se deforman en una superficie hidrofóbica: las gotas ajustan su forma para minimizar el contacto con una superficie hidrofóbica.
Las moléculas de agua adyacentes a un hidrocarburo mantienen interacciones moleculares con otras moléculas de agua y, al hacerlo, se ve reducida su entropía. Las fuertes interacciones cohesivas direccionales entre las moléculas de agua se mantienen, pero a un alto coste entrópico. La baja entropía del agua en la región interfacial surge de las fuertes fuerzas direccionales entre las moléculas de agua. En agua en forma líquida, estas fuerzas son esencialmente isotrópicas (se extienden en todas las direcciones). En solución, una molécula de agua puede rotar y al mismo tiempo mantener interacciones de enlace de hidrógeno. En una interfaz hidrofóbica, las interacciones son anisotrópicas (direccionales) porque el hidrocarburo no forma enlaces de hidrógeno. De este modo, el componente entrópico resulta en una energía libre desfavorable de mezcla entre aceite y agua (ΔG = ΔH-TΔS> 0).
El término "enlace hidrofóbico" es un concepto inapropiado y debe evitarse, aunque Walter Kauzmann, el descubridor del efecto hidrofóbico, lo usó con frecuencia.
La descripción anterior del efecto hidrofóbico a nivel molecular puede entenderse mediante los parámetros termodinámicos de entalpía (ΔH, indica cambios en las interacciones moleculares) y entropía (ΔS, indica cambios en los estados disponibles de rotación, traslación, vibración, etc.). Un hidrocarburo mantiene interacciones moleculares favorables con las moléculas de agua en una solución acuosa. Sabemos esto porque la transferencia de un mol de hidrocarburo puro a una solución acuosa diluida tiene una entalpía de alrededor de cero. Entonces, ¿por qué no se mezclan el aceite y el agua? El responsable es el agua. El agua expulsa las sustancias no polares de la fase acuosa.
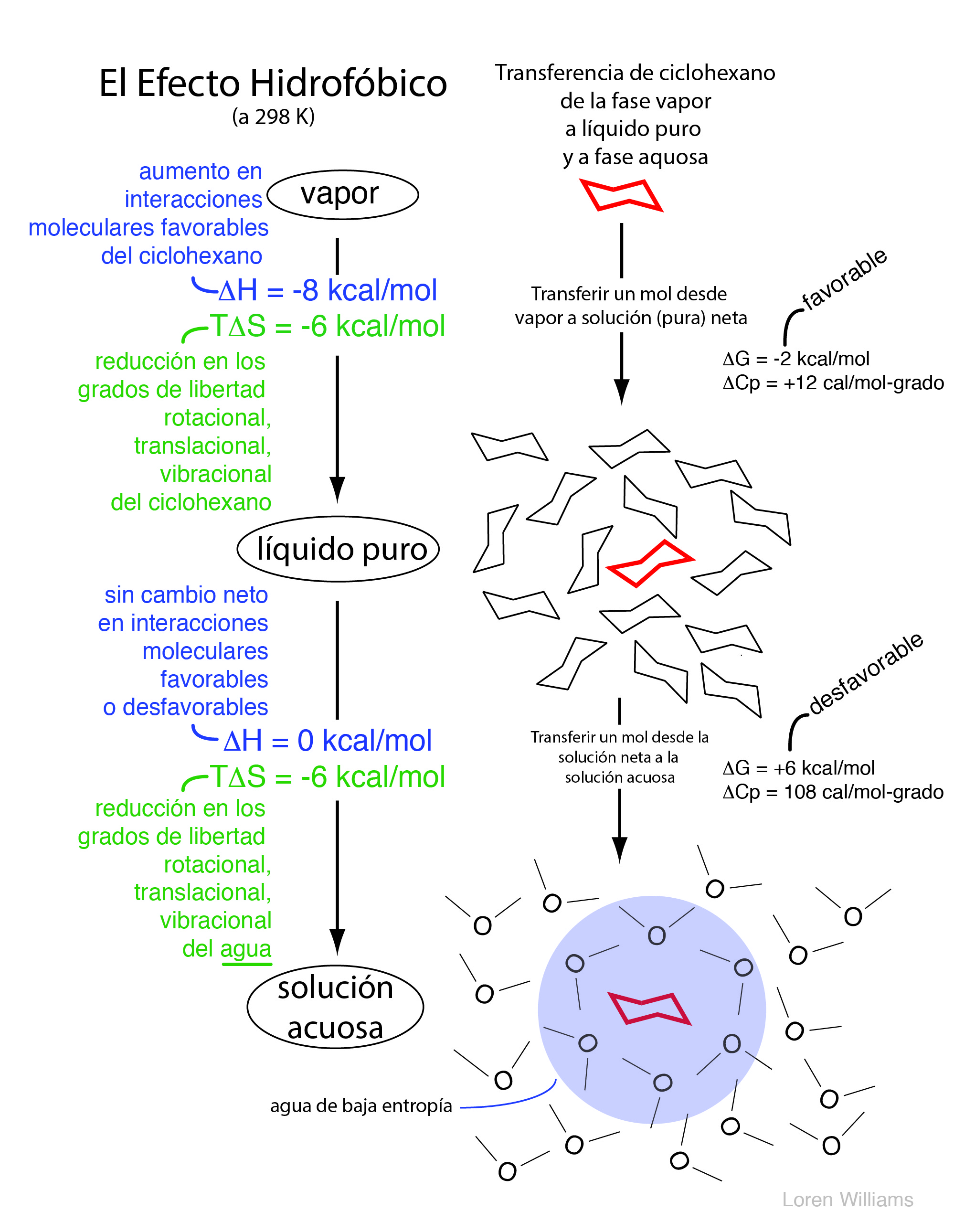
La figura 28 ilustra lo que sucede cuando una sustancia hidrófoba (ciclohexano en este caso) se convierte de vapor a líquido puro y luego a solución acuosa. En el primer paso, pasando de la fase de vapor al líquido puro, hay un aumento en las interacciones intramoleculares y una disminución en los grados de libertad rotacional y translacional. Por lo tanto se espera, y se observa, una contribución de entalpía favorable (ΔH negativa) y una contribución de entropía desfavorable (TΔS negativa) para la condensación. En el segundo paso, pasando de líquido puro a solución acuosa diluida, el cambio en la estabilidad contribuido por las interacciones intramoleculares es nulo. La entalpía de transferencia es cercana a cero. Pero el agua pierde entropía. El agua está más ordenada en las proximidades de una molécula de ciclohexano que en agua pura. Por lo tanto, para este paso, ΔH es cero, TΔS es negativo y ΔG es positivo (ΔG = ΔH-TΔS).
Como se ilustra a continuación, en la fase acuosa se forma una región de relativamente baja entropía (alto orden) en la interfaz entre el solvente acuoso y un soluto hidrofóbico.
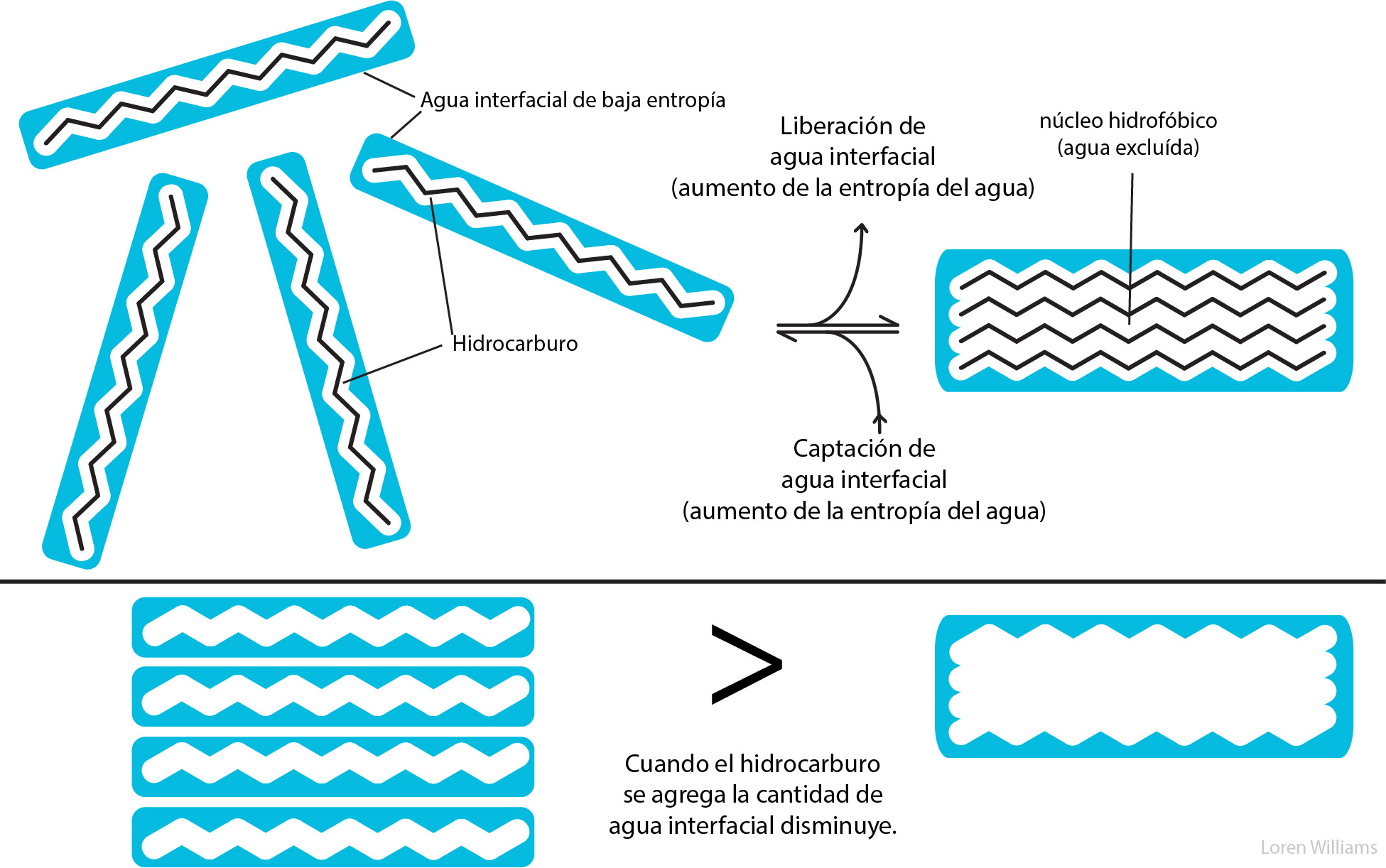
La figura 29 muestra cómo la agregación de moléculas de hidrocarburos provoca la liberación de moléculas de agua interfaciales. Por lo tanto, el sistema gana entropía (TΔS positivo) tras la agregación de los hidrocarburos. La liberación de moléculas de agua interfacial de baja entropía a la solución impulsa la agregación de hidrocarburos. El panel inferior ilustra que hay más agua interfacial en el lado izquierdo de la ecuación que en el lado derecho.
Cuando las moléculas de hidrocarburo se agregan en solución acuosa, el volumen total de agua interfacial disminuye. Por lo tanto, la fuerza impulsora de la agregación de sustancias hidrofóbicas es el resultado de un aumento en la entropía del agua. La fuerza impulsora para la agregación no surge de la atracción intrínseca entre las moléculas hidrofóbicas de soluto.
Si consideramos la entropía de las moléculas de hidrocarburos de forma independiente, una solución dispersa tiene mayor entropía y es más estable que un estado agregado. De manera similar, una proteína puede parecer tener una mayor entropía en un ovillo aleatorio que en un estado nativo. Solo cuando la entropía de la fase acuosa se tiene en cuenta en la ecuación se puede entender la separación de agua y aceite en dos fases, y el plegamiento de una proteína en un estado nativo.
Para muchos propósitos, es útil considerar al ADN como una barra recubierta con carga aniónica. En solución acuosa, la barra negativa está rodeada de contraiones (cationes como Na+, K+ y Mg2+ y/o poliaminas). La liberación de los contraiones explica gran parte de la dependencia de las sales para la desnaturalización de ADN, las interacciones de ADN-proteína, el plegamiento de ARN y la condensación de ADN.
La alta densidad de carga negativa en la barra provoca fuertes campos eléctricos radiales. El campo eléctrico es fuerte cerca de la barra y débil lejos de ella. Estos campos eléctricos conducen a la formación de pronunciados gradientes radiales en la concentración de contraiones. La concentración de contraiones es alta cerca de la barra y baja lejos de ella. Los contraiones "condensados" son móviles, pero están limitados a un pequeño volumen en las proximidades del ADN.
Los modelos teóricos muestran que la concentración local de un catión monovalente como K+ cerca de la superficie del ADN es de aproximadamente 2 molar. Las razones no son obvias, pero la concentración de K+ que rodea el ADN es en gran medida independiente de la concentración de K+ en el medio acuoso. El entorno electrostático que rodea el ADN no depende de la concentración de los contraiones del medio acuoso en general.
Desnaturalización de ADN. Cuando el ADN se deshibrida, las hebras se separan. La separación de filamentos libera contraiones condensados.
ADN dúplex → cadena 1 + cadena 2 + contraiones libres
Esta relación explica por qué aumenta la estabilidad del ADN bicatenario (con una Tm más alta) a medida que aumenta la concentración de las sales (fuerza iónica). La aplicación del principio de Le Chatelier muestra que la adición de contraiones empuja el equilibrio hacia la izquierda, hacia la formación del dúplex.
Interacciones Proteína-ADN. Se liberan contraiones cuando una proteína catiónica se añade al ADN.
ADN + proteína → ADN-proteína + contraiones libres
Una alta concentración de sal desestabiliza los complejos ADN-proteína. La liberación de iones explica esta dependencia de las sales. La aplicación del principio de Le Chatelier muestra que la adición de contraiones empuja el equilibrio hacia la izquierda, hacia el ADN y la proteína disociados.
Si la cantidad de sales es baja, hay una gran ganancia entrópica de la liberación de contraiones, y la proteína se adhiere fuertemente al ADN. Si la cantidad de sales es alta, la ganancia entrópica de la liberación de contraiones es pequeña, y la proteína se adhiere débilmente al ADN.
Condensación de ADN. Los ADN genómicos son moléculas muy largas. Los 160.000 pares de bases de ADN del fago T4 se extienden hasta 54 micras. Los 4.2 millones de pares de bases del cromosoma de E. coli se extienden hasta 1.4 milímetros. En los sistemas biológicos, las moléculas de ADN deben compactarse para que ocupen espacios muy pequeños dentro de una célula, núcleo o partícula de virus. Las barreras energéticas para el empaquetamiento compacto del ADN surgen de la disminución de la entropía configuracional, la flexión de la doble hélice rígida y la repulsión electrostática intermolecular (o entre segmentos) de los grupos fosfato de ADN con carga negativa. Sin embargo, las cadenas de ADN extendidas se condensan espontáneamente por colapso en partículas muy compactas y muy ordenadas. En el estado condensado, las hélices de ADN están separadas por una o dos capas de agua. Las partículas de ADN condensadas toman comúnmente la forma de toroides compactos. La condensación de ADN en solución acuosa requiere cationes altamente cargados como la espermina (+4) o la espermidina (+3). Los cationes divalentes condensarán el ADN en mezclas de agua y alcohol. El papel de los cationes es disminuir la repulsión electrostática de segmentos adyacentes de ADN con carga negativa. La fuente de la atracción entre segmentos de ADN cercanos no es tan fácil de entender. Una posible fuente de atracción son las fluctuaciones de las atmósferas iónicas en analogía con los dipolos fluctuantes entre las moléculas (Fuerzas de London).
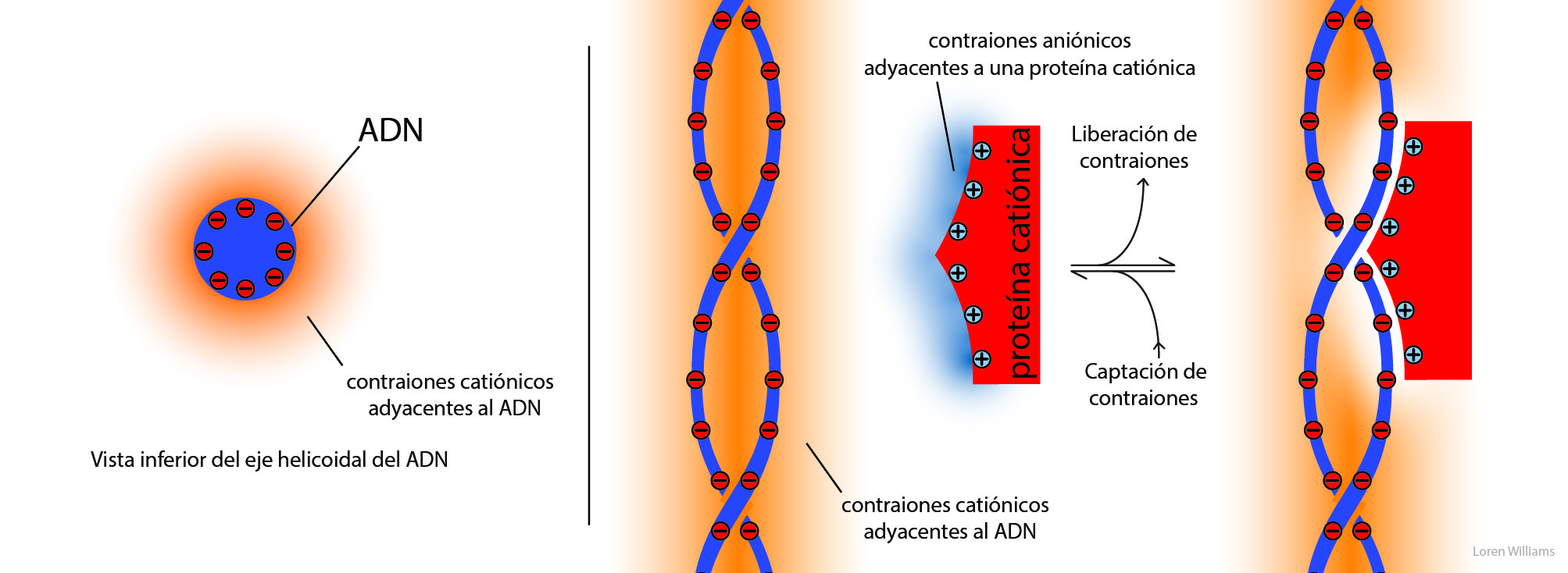
La figura 30 (izquierda) muestra una vista axial del ADN, representado como un cilindro aniónico. Los contraiones catiónicos (contorno anaranjado) rodean el cilindro. La concentración de cationes disminuye con la distancia desde la superficie del cilindro. El sombreado anaranjado más intenso indica una mayor concentración de cationes. El panel de la derecha ilustra cómo los contraiones aniónicos (azul) asociados a una proteína catiónica y los contraiones catiónicos (naranja) asociados al ADN aniónico, se liberan al entorno cuando la proteína se une al ADN. Esta liberación de contraiones impulsa la asociación (contribuyendo + TΔS).
Este documento está dedicado a la memoria del difunto profesor Charles Lochmuller (derecha) de la Universidad de Duke. El Dr. Lochmuller era un buen hombre, un cómico natural y un eminente científico.
Aquí me acerco a la bioquímica de una manera nueva (eso creo). Es tradición comenzar con el primer libro de texto de Bioquímica de Lehninger y continuar esencialmente con todos los libros de texto de bioquímica posteriores, para tratar cada tipo de biopolímero de forma aislada de los demás. Proteínas, ADN, ARN y carbohidratos se describen en capítulos distintos y bien separados como fenómenos químicos no relacionados.
Tuve la suerte de aprender interacciones moleculares del Dr. Lochmuller en su clase de separaciones. He ampliado el esquema de análisis del Dr. Lochmuller para incluir catión-Π, etc. Parte del material utilizado para la Parte 1 del documento proviene de mi tesis doctoral (1984). Escribí elementos centrales entre 1990-92, y expando, reviso y clarifico las figuras y el texto cuando estoy inspirado y tengo tiempo disponible. El documento se ha beneficiado de muchas discusiones con el profesor Nicholas Hud. Creé este recurso y continúo mejorándolo en base a mis creencias de que:
El desarrollo de este documento ha sido apoyado por el Instituto de Astrobiología de la NASA, la Fundación Nacional de Ciencias (NSF) y la Escuela de Química y Bioquímica de Georgia Tech, los cuales han apoyado mi laboratorio de investigación y mis esfuerzos de divulgación científica. Los comentarios y sugerencias de mejoras para la versión en español son bienvenidos y deben dirigirse a celiablanco@ucla.edu o igallego@mrc-lmb.cam.ac.uk. Espero que los estudiantes, especialmente aquellos que carecen de recursos para libros de texto, encuentren este sitio útil.
Uso. Las imágenes y el texto aquí se pueden reutilizar con atribución para fines no comerciales.
Sinceramente,
Loren Williams,
Professor,
Georgia Tech
ldw@gatech.edu
